Lékařský expert článku

Nové publikace
hamartom
Naposledy posuzováno: 29.06.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
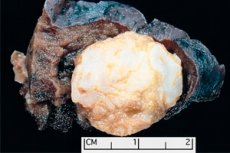
Nádorovitý útvar lokalizovaný v jakékoli anatomické oblasti, vzniklý abnormálním růstem benigní tkáně, je v medicíně definován jako hamartom (z řeckého hamartia - chyba, vada). [ 1 ]
Epidemiologie
Statisticky představují hamartomy 1,2 % benigních novotvarů. Prevalence plicních hamartomů se odhaduje na přibližně 0,25 % běžné populace a představuje až 8 % všech plicních novotvarů. Většina plicních hamartomů je diagnostikována náhodně u pacientů ve věku 40 až 70 let, ale v pediatrické praxi jsou velmi vzácné.
Obecně je většina hamartomů diagnostikována u mužů, ačkoli v ledvinách jsou častější u žen a jsou identifikovány ve středním věku.
Asi 5 % benigních nádorů prsu jsou hamartomy a nejčastěji postihují ženy starší 35 let.
80–90 % hamartomatózních lézí mozku a více než 50 % hamartomů srdce je spojeno s tuberózní sklerózou.
Příčiny hamartomy
Gamartomy patří mezi vrozené vady a jsou to útvary benigního charakteru, které vznikají z mezenchymálních tkání pocházejících ze zárodečných listů. A příčiny jejich výskytu jsou spojeny s nekontrolovaným buněčným dělením cytologicky normálních tkání (pojivové, hladké svaloviny, tukové nebo chrupavky), charakteristických pro danou anatomickou lokalizaci, a jejich ložiskovým přerůstáním během embryogeneze téměř jakéhokoli orgánu nebo anatomické struktury.
Výskyt více hamartomů u stejného pacienta se často označuje jako hamartomatóza nebo pleiotropní hamartom.
Tyto nádory se mohou vyskytovat sporadicky nebo v přítomnosti určitých autozomálně dominantně dědičných onemocnění, stejně jako geneticky podmíněných syndromů.
V mnoha případech se hamartomy tvoří, když se krátce po narození projeví vzácné genetické onemocnění multisystémové povahy – tuberózní skleróza – nebo při Recklinghausenově familiární chorobě – neurofibromatóze typu 1. [ 2 ]
Rizikové faktory
Mezi hlavní rizikové faktory pro vznik hamartomu patří přítomnost tzv. genetických syndromů hamartomatózní polypózy v anamnéze pacienta, včetně:
- Syndrom mnohočetných hamartomů - Cowdenův syndrom, při kterém se tvoří mnohočetné hamartomy ekto-, ento- a mezodermálního původu, pozorují se gastrointestinální polypózy a mukokutánní projevy;
- Peutz-Jeghers-Turenův syndrom (charakterizovaný vývojem benigních hamartomatózních polypů v gastrointestinálním traktu);
- Proteův syndrom;
- Weilův syndrom - juvenilní polypóza tlustého střeva;
- Bannayan-Riley-Ruvalcabův syndrom, který stejně jako Cowdenův syndrom způsobuje mnohočetné hamartomy (hamartomatózní polypy) střeva;
- Carney-Stratakisův syndrom a Carneyho komplex.
Kromě toho se hamartomy tvoří u pacientů s hereditárním Watsonovým syndromem a v případech sporadicky se vyskytujícího nebo vrozeného Pallister-Hallova syndromu s hypotalamickým hamartomem a polydaktylií.
Patogeneze
Mechanismus zvýšené proliferace zárodečných tkání s tvorbou nádorovitých malformací v různých orgánech je vysvětlen chromozomálními aberacemi a genovými mutacemi, které mohou vznikat spontánně nebo být zděděné.
U tuberózní sklerózy byly identifikovány mutace v genech TSC1 nebo TSC2 – tumor supresorových genech, které zabraňují a inhibují nadměrnou proliferaci – příliš rychlý nebo nekontrolovaný růst a dělení buněk. A u neurofibromatózy typu 1 a Watsonova syndromu – zárodečné mutace mitochondriálního tumor supresorového genu NF1.
U syndromu hamartomového tumoru, který kombinuje Cowdenův, Proteův, Bannayan-Riley-Ruvalcabův syndrom a syndrom juvenilní polypózy, je patogeneze spojena s mutací genu PTEN, který kóduje enzym zapojený do regulace proliferace a je považován za tumor supresorový gen.
Mutace v genu STK11 kódujícím strukturu a funkci jednoho z transmembránových serinových enzymů, které snižují jeho schopnost omezovat buněčné dělení, vedou k Peutz-Jeghers-Turenovu syndromu s rozvojem střevních polypů a pigmentovaných kožních lézí. U Pallister-Hallova syndromu byla identifikována mutace v genu GLI3, transkripčním faktoru zapojeném do tvorby nitroděložní tkáně.
Nekontrolovaný růst buněk v důsledku genových mutací tedy vede k tvorbě hamartomů.
Symptomy hamartomy
V závislosti na lokalizaci hamartomů se rozlišují jejich typy a každý z nich má svou vlastní strukturu a symptomatologii.
Hamartom plic
Plicní hamartom se může tvořit v jakémkoli laloku a periferních částech plic a skládá se z normálních tkání přítomných v plicích: tukové, epiteliální, fibrotické a chrupavčité. V 80 % případů převládá chondroidní složka (hyalinní chrupavčité buňky) se zahrnutím adipocytů - buněk tukové tkáně a epiteliálních buněk dýchacích cest. [ 3 ]
Dřívější názvy: chondroidní hamartom, mezenchymom, chondromatózní hamartom nebo hamartochondrom v současné době WHO nedoporučuje.
Mezenchymální cystický hamartom plic je naopak méně častý a u většiny pacientů je spojen s Cowdenovým syndromem.
Hamartomatózní léze plic se nemusí projevovat, ale může způsobovat příznaky ve formě chronického kašle (často s hemoptýzou), sípání při dýchání a dýchacích potíží. [ 4 ]
Hamartom srdce
Mezi benigní primární srdeční nádory u dospělých patří hamartom zralých myocytů a u kojenců a dětí s tuberózní sklerózou rabdomyom, tj. hamartom myokardu komor nebo mezikomorového septa. [ 5 ]
Zralý kardiomyocytární hamartom se vyvíjí ve stěně komor (a vzácně v síních) a může se projevovat jako mnohočetné léze, které jsou hustými masami úzce spojenými s podkladovým myokardem. Nádor může způsobovat příznaky srdečního selhání: bolest na hrudi, palpitace a arytmie, srdeční šelesty, otoky, dušnost, cyanózu.
Srdeční rabdomyomy, z nichž většina je diagnostikována v prvním roce života, se skládají z tkáně srdečního svalu tvořené embryonálními myoblasty a mají vzhled pevných ložiskových mas bez pouzdra.
Tyto hamartomy se obvykle projevují asymptomaticky a spontánně ustupují před dosažením věku 4 let.
Hamartomatózní léze jsou některými odborníky také považovány za spojované s myxomem srdce Carneyho komplexu. [ 6 ]
Gamartom gastrointestinálního traktu
Gastrický hamartom je mezenchymální masa ve formě epiteliálního hyperplastického polypu žaludku, Peutz-Jeghersova polypu a vzácného myoepiteliálního hamartomu - s hypertrofovanými svazky hladkého svalstva. Další názvy tohoto hamartomu zahrnují myoglandulární hamartom, adenomyomatózní hamartom a gastrický adenomyom. Mezi typické klinické projevy patří dyspepsie, bolest v epigastriu a krvácení do horní části gastrointestinálního traktu. [ 7 ], [ 8 ]
Více informací v materiálu - žaludeční polypóza
Střevní hamartom je hamartomatózní nebo hyperplastický polyp tlustého střeva, diagnostikovaný jako adenomatózní nebo tubulární adenom. Pokud je hamartom lokalizován v Brunnerově žláze dvanáctníku, příznaky se projevují bolestí v epigastrické oblasti, nevolností, zvracením a plynatostí (což naznačuje střevní obstrukci); a pokud je hamartom značné velikosti, gastrointestinálním krvácením. V případech myoepiteliálního hamartomu ilea si pacienti stěžují na bolesti břicha, mají sníženou tělesnou hmotnost a rozvíjí se u nich chronická anémie. [ 9 ], [ 10 ]
Čtěte také - rektální polypy
Retrorektální hamartom je cystický hamartom neboli vícekomorová cysta retrorektálního prostoru (řídké pojivové tkáně mezi konečníkem a vlastní fascií), která se nejčastěji vyskytuje u žen středního věku. Má vzhled cysty vyčnívající ze zadní stěny konečníku, která je vystlána epitelem a obsahuje chaoticky uspořádaná vlákna hladkého svalstva. Tento hamartom se projevuje bolestmi v podbřišku a opakující se zácpou. [ 11 ], [ 12 ]
Hamartomy jater a sleziny
Mnohočetný biliární hamartom jater je hamartom interdolických intrahepatálních žlučovodů spojený s malformacemi jejich vývoje během embryonálního období. Tento hamartom (jednoduchý nebo mnohočetný) se skládá z nahodile rozšířených shluků žlučovodů a fibrokolagenního stromatu. [ 13 ]
Biliární hamartomy jsou asymptomatické a obvykle se objevují náhodně (během radiologického vyšetření nebo laparotomie). [ 14 ]
Vzácným a často náhodně zjištěným primárním nádorem benigního charakteru je hamartom sleziny, který se skládá z prvků červené dřeně sleziny - ve formě dobře definované homogenní masy pevné konzistence. Tato malformace může být jednoduchá nebo vícečetná; při stlačení parenchymu sleziny se může objevit pocit diskomfortu a bolesti v levé subkostální oblasti. [ 15 ], [ 16 ]
Renální hamartomy
Nejčastějším hamartomem ledviny je diagnostikován angiomyolipom ledviny, protože tento benigní nádor se skládá ze zralé tukové tkáně s vloženými vlákny hladkého svalstva a krevními cévami. Vzniká při tuberózní skleróze ve 40–80 % případů. Zvětšení velikosti hamartomu (více než 4–5 cm) vede k bolesti a výskytu krve v moči. [ 17 ], [ 18 ]
Hamartom prsu
Diagnostické definice hamartomu prsu, které uznávala WHO, zahrnují termíny jako adenolipom, chondrolipom a myoidní hamartom. Ačkoli je mamologové často označují jako fibroadenolipom, protože nádorový útvar obsahuje buňky fibrotické, žlázové a tukové tkáně uzavřené v tenkém pouzdře pojivové tkáně s výraznými obrysy. Při vizualizaci lze pozorovat ložiskové kalcifikace. V tomto případě klinické projevy chybí. [ 19 ], [ 20 ]
Čtěte také - Nádory prsu
Hamartomy mozku
Třetina pacientů s tuberozní sklerózou má mozkový hamartom ve formě intrakraniálních kortikálních výrůstků nebo tuberkul v různých lalocích - na hranici šedé a bílé hmoty - nebo subependymálních uzlíků podél stěn mozkových komor. Může se také tvořit astrocytický hamartom, subependymální obrovskobuněčný astrocytom s kortikální disrupcí, dysmorfními neurony a velkými gliovými buňkami mozkového parenchymu (astrocyty). Mezi příznaky mozkových hamartomů patří záchvaty a mentální retardace u dětí. [ 21 ], [ 22 ]
Vzácnou malformací, která se vyskytuje během embryogeneze a je přítomna již při narození, je hypotalamický hamartom, což je masa heterotopních neuronů a gliových buněk. S růstem mozku dítěte se nádor zvětšuje, ale nešíří se do dalších oblastí mozku. [ 23 ], [ 24 ]
Pokud se v přední části hypotalamu (tuber cinereum), kde se na něj napojuje hypofýza, vytvoří hypertrofované tkáně, projevuje se malformace příznaky centrálního předčasného pohlavního vývoje (před 8.–9. rokem věku): výskyt akné, časný vývoj mléčných žláz a časná menstruace u dívek; časné ochlupení a hlasová mutace u chlapců.
Pokud se hamartomy tvoří v zadní části hypotalamu, mohou se objevit abnormality v elektrické aktivitě mozku, které se v raném kojeneckém věku projevují záchvaty a v pozdějším stádiu (ve věku 4 až 7 let) epilepsií s fokálními epileptickými záchvaty s náhlým smíchem nebo s mimovolním pláčem, atonickými a tonicko-klonickými záchvaty, ale i záchvaty agrese, poruchami paměti a kognitivními problémy.
Hamartom hypofýzy je sporadicky se vyskytující benigní adenom hypofýzy.
Dospělí středního věku s Cowdenovým syndromem mohou mít vzácný nádorovitý útvar, hamartom mozečku, diagnostikovaný jako dysplastický mozečkový gangliocytom nebo Lhermitte-Duclosova choroba. Příznaky mohou chybět nebo se projevovat jako bolest hlavy, závratě, zhoršená koordinace pohybů a paralýza jednotlivých hlavových nervů.
Hamartom lymfatické uzliny
Když dochází k přemnožení buněk hladkého svalstva a tukové tkáně, stejně jako krevních cév a kolagenního stromatu tříselných, retroperitoneálních, submandibulárních a krčních lymfatických uzlin, vzniká angiomyomatózní hamartom lymfatické uzliny nebo nodulární angiomyomatózní hamartom - s částečnou nebo úplnou náhradou jejího parenchymu. [ 25 ], [ 26 ]
Hamartom kůže
V přítomnosti tuberózní sklerózy nebo neurofibromatózy se pozorují různé hamartomy kůže, nejčastěji ve formě hypopigmentovaných skvrn; kávových a mléčných skvrn; angiofibromu (na tvářích, bradě, nasolabiálních záhybech); shagreenových skvrn různých lokalizací (což jsou névy pojivové tkáně); vláknitých plaků na čele, pokožce hlavy nebo krku.
Vzácným dermatologickým projevem tuberózní sklerózy (zejména u mužů) je folikulocystický a kolagenní hamartom, charakterizovaný hojným ukládáním kolagenu v dermis, koncentrickou perifolikulární fibrózou a keratinem vyplněnými trychtýřovitými subkutánními cystami, které jsou patrné při histopatologickém vyšetření. [ 27 ]
K hamartomům sestávajícím z melanocytů (buňek produkujících pigment melanin) většina odborníků označuje také různé melanocytární neoplazmy, zejména vrozené melanocytární névy, které představují abnormalitu embryogeneze.
Z hlediska etiologie jsou hamartomy složené z cévní tkáně také hemangiomy kůže.
Pacienti s Peutz-Jeghers-Thurenovým syndromem mají hamartom ve formě skvrnité pigmentace kůže a sliznic - lentiginosis periorificialis
Případy lineárního papulárního ektodermálně-mezodermálního hamartomu (Hamartoma moniliformis) vykazují lineární papulární vyrážku tělové barvy na hlavě, krku a horní části hrudníku.
A sebocytární hamartom je hamartom mazových žláz, více si přečtěte v publikaci - mazový névus.
Hamartom oka
Pigmentované hamartomatózní léze duhovky u neurofibromatózy typu 1 a Watsonova syndromu - ve formě nodulárních shluků dendritických melanocytů - jsou definovány jako hamartomy duhovky nebo Lischovy uzlíky. Jsou to průhledné (obvykle neovlivňující vidění) zaoblené kopulovité žlutohnědé papuly, které vyčnívají nad povrch duhovky.
A u pacientů s juvenilním angiofibromem nosohltanu a familiární adenomatózní polypózou se často vyvine kombinovaný hamartom sítnice a retinálního pigmentového epitelu – ve formě černé skvrny na centrální (makulární) části sítnice. [ 28 ]
Hamartom nosu
Nosní hamartom je specialisty definován jako nosní chondromesenchymální hamartom nebo nosní chondrom, způsobený benigní proliferací respiračního epitelu, submukózních žláz a chondro-kostního mezenchymu. Jeho klinické projevy závisí na velikosti a lokalizaci léze a zahrnují: ucpaný nos, potíže s dýcháním nosem a kojením u kojenců, čirý vodnatý výtok z nosu a krvácení z nosu. Hamartom může růst s dítětem a šířit se do očních orbity, což vede k posunu oční bulvy dopředu nebo dozadu, strabismu nebo poruchám okulomotoriky. [ 29 ]
Hamartom u dítěte
Všechny výše uvedené hamartomatózní léze různých orgánů a anatomických struktur jsou přítomny u dětí s odpovídajícími syndromy.
Novorozenci se projevují mezenchymálním hamartomem hrudní stěny nebo chrupavčitým hamartomem žebra, což jsou pevné nepohyblivé masy vzniklé v důsledku fokálního přerůstání normálních kosterních elementů chrupavčitými, cévními a mezenchymálními prvky. Tento hamartom může způsobit respirační selhání a rozvoj syndromu respirační tísně. Mezenchymální hamartom jater je druhým nejčastějším benigním nádorem jater u dětí. Tento nádorovitý útvar (častěji lokalizovaný v pravém laloku orgánu) se skládá z buněk mezenchymálního stromatu, hepatocytů a epiteliálních buněk výstelky žlučovodů. Klinický obraz zahrnuje hmatatelnou masu v břišní dutině, nechutenství a úbytek hmotnosti a v případě významné velikosti (až 10 cm a více) nádor pokrývá extrahepatální žlučovody a dolní dutou žílu, což vede k žloutence a edému dolních končetin.
Hamartom je vrozený mezoblastický nefrom (vyskytuje se u 1 z 200 000 kojenců), který může u novorozence vést k nadýmání břicha s hmatatelnou masou husté konzistence v pravém horním kvadrantu břicha. Kojenci se také mohou projevovat zrychleným mělkým dýcháním.
Mezi vzácné vrozené anomálie patří fibrotický hamartom v kojeneckém věku, který se vyskytuje u dětí v prvních dvou letech života a projevuje se jako bezbolestná uzlovitá masa v podkožních tkáních axily, krku, ramene a předloktí, zad a hrudníku, stehna, chodidla a zevních genitálií.
Ekrinní angiomatózní hamartom u dítěte může být přítomen již při narození nebo se projevit v raném dětství. Tento benigní nádor hamartomatózní povahy má obvykle vzhled modravých nebo nahnědlých uzlíků a/nebo plaků, které vznikají proliferací tkáně ekrinních potních žláz a kapilár ve středních a hlubokých vrstvách dermis. Tento hamartom může způsobit lokalizovaný hyperhidrózu a zvýšený růst ochlupení.
Komplikace a důsledky
Panuje všeobecná shoda, že hamartomy se jen zřídka vracejí nebo transformují do maligních nádorů. Často vykazují jen malé nebo žádné příznaky a někdy dokonce časem mizí. V závažnějších případech a v závislosti na místě vzniku však mohou mít tyto malformace závažné komplikace a následky.
Za prvé, hamartom může dorůst do takové velikosti, že začne tlačit na okolní tkáně a orgány a narušovat jejich funkce.
Srdeční hamartom u dětí může vést k přetrvávajícím abnormalitám srdečního rytmu, chlopenním vadám a zhoršenému intrakardiálnímu průtoku krve s následným městnavým srdečním selháním.
Komplikace hamartomatózních polypů gastrointestinálního traktu zahrnují gastrointestinální krvácení, obstrukci a střevní invaginaci (s možným fatálním koncem). A velký ledvinový hamartom může vyvolat rupturu ledviny.
Hamartom v mozku může způsobit syndrom obstrukčního hydrocefalu.
U hamartomů hypotalamu a hypofýzy může být narušena produkce somatotropního hormonu (růstového hormonu), což vede k rozvoji hypofyzeálního nanismu (hypopituitarismu) u dětí. Hamartomy hypotalamu u dětí mohou také vést k epilepsii rezistentní na léky.
Komplikace hamartomu pigmentového epitelu sítnice jsou plné dysfunkce sítnice a/nebo zrakového nervu, makulárního edému, neovaskularizace cévnatky a odchlípení sítnice.
Diagnostika hamartomy
Důležitou součástí diagnostiky hamartomů a souvisejících syndromů je sběr anamnézy, včetně rodinné anamnézy.
Laboratorní testy zahrnují krevní testy: obecné klinické; sérové elektrolyty; lymfocytární profil; hladiny vápníku, draslíku, fosfátů a močoviny; a jaterní testy. Pokud je to možné, provádí se tenkojehlová aspirační punkční biopsie útvaru, protože histologické vyšetření je klíčové pro diagnózu a volbu léčebné taktiky.
Instrumentální diagnostika zajišťuje vizualizaci hamartomatózního tumoru podobného útvaru a identifikaci jeho přesné lokalizace, k čemuž se využívá rentgen, angiografie, elektroencefalografie (EEG), ultrazvuk (sonografie), CT (počítačová tomografie), PET (pozitronová emisní tomografie), MRI (magnetická rezonance).
Diferenciální diagnostika
U jakýchkoli abnormálních nádorů je diferenciální diagnostika velmi důležitá. Proto se rozlišuje tuberkulom a hamartom; plicní hamartom a primární karcinom plic, bronchogenní karcinoid, metastatické onemocnění. Mozkový hamartom je třeba odlišit od kraniofaryngeomu a hypotalamo-chiasmatického gliomu. Diferenciální diagnostika hamartomu jako vrozeného mezoblastického nefromu zahrnuje Wilmsův tumor (maligní nefroblastom), světlobuněčný sarkom ledvin a osifikující tumor ledvin u kojenců.
Kdo kontaktovat?
Léčba hamartomy
Pokud je hamartom asymptomatický a je objeven náhodně, není nutná žádná léčba, ale je nutné sledovat jeho „chování“ a stav pacienta. V jiných případech je terapie zaměřena na snížení intenzity symptomů a prevenci komplikací. Například u hypotalamického hamartomu s příznaky předčasné puberty se předepisují určité léky, které inhibují uvolňování určitých hormonů. Kardioléky se používají k léčbě symptomů srdečního selhání u pacientů se srdečními hamartomy.
Chirurgické odstranění hamartomů je indikováno k potvrzení diagnózy a v případech lékařsky nekorigovatelných intenzivních symptomů.
Například plicní hamartomy lze resekovat klínovou resekcí a v závažných případech odstraněním laloku plic (lobektomie). Lze také odstranit hamartom prsu a pokud je velký, může být nutná částečná nebo úplná mastektomie.
K odstranění hamartomatózních polypů lze použít stereotaktickou radiofrekvenční termoablaci nebo laserovou ablaci. Používá se také radiochirurgie s vysoce fokusovanými gama paprsky - gama nůž pro hypotalamické hamartomy nebo astrocytické hamartomy.
Prevence
Jedinou metodou prevence vzniku hamartomů lze považovat genetické vyšetření budoucích rodičů dítěte.
Předpověď
Celková prognóza této vrozené anomálie závisí na lokalizaci a velikosti novotvaru, stejně jako na komorbiditách a celkovém zdravotním stavu pacienta.