Lékařský expert článku

Nové publikace
Vrozené aneurysma
Naposledy posuzováno: 29.06.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
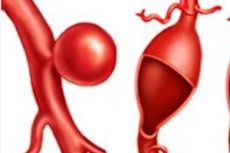
Patologické oslabení a následné lokalizované vyboulení stěny arteriální cévy, srdeční komory nebo mezisíňové přepážky, ke kterému dochází v důsledku vrozené vady nebo genetického onemocnění, se diagnostikuje jako vrozené aneurysma.
Epidemiologie
Syndromální aneuryzmata tvoří 10–15 % případů; aneuryzmata a disekce hrudní aorty se vyskytují u 80 % pacientů s Marfanovým syndromem.
Podle studií je prevalence aneuryzmat plicní tepny u vrozených srdečních vad asi 6 % případů a prevalence vrozených aneuryzmat levé komory u dospělých nepřesahuje 0,7 %. Aneuryzmata síňového septa se vyskytují u 1 % dětí a 1–2 % dospělých.
Výskyt aneuryzmatu aortálního sinu se odhaduje na 0,09 % v běžné populaci a představuje 3,5 % všech vrozených srdečních vad.
Mozkové aneuryzmata se vyvíjejí přibližně u 50 % pacientů s vrozenými arteriovenózními malformacemi mozku, ale u dětí mladších 12 let tvoří méně než 5 % případů. Podle klinických statistik se 85 % všech intrakraniálních aneuryzmat vyskytuje v cévách Willisova kruhu v mozku.
Příčiny vrozené aneurysma
Odborníci berou v úvahu příčiny vrozených aneuryzmat a kladou na první místo geneticky podmíněné patologie pojivové tkáně:
- Marfanův syndrom;
- Ehlers-Danlosův syndrom (vaskulární typ);
- Loesův-Dietzův syndrom.
Arteriální aneuryzmata se snadněji tvoří u pacientů s Williamsovým syndromem (Williams-Boyren), který je důsledkem delece genu ELN, jenž kóduje protein tropoelastin, prekurzor elastinu (důležitý strukturní prvek extracelulární matrix tkání cévní stěny).
Definovány jsou také nesyndromální hereditární (familiární) aneuryzmata hrudní aorty a již byly identifikovány mutace v některých genech (MYH11, ACTA2 atd.).
Aneuryzmata vrozeného původu se mohou tvořit v přítomnosti genetických enzymatických abnormalit - různé typy mukopolysacharidózy u dětí, které vznikají mutacemi v genech kódujících enzymy potřebné k metabolismu glykosaminoglykanů (mukopolysacharidů).
Mozkové aneurysmata přítomná při narození nejsou spojena pouze se syndromálními kolagenózami. Takovéto fokální vyboulení stěn mozkových cév v oblasti mozkového arteriálního kruhu, Willisiova kruhu, se pozoruje u:
- Arteriovenózní malformace - vrozená cévní patologie, jejíž podstatou je porušení architektoniky arteriálních a žilních cév, což vede ke změnám v normálním průtoku krve;
- Autozomálně dominantní polycystické onemocnění ledvin.
Vrozené aneurysma břišní aorty nebo renálních tepen může být neurofibromatóza typu 1, genetická porucha, která je výsledkem spontánní mutace u téměř poloviny pacientů.
Za příčiny žilních aneurysmat (které jsou v klinické praxi vzácné) se považují cévní malformace vzniklé během nitroděložního vývoje (např. u Servel-Martorellova syndromu), dále žilní angiodysplazie typu žilní dilatace u Klippel-Trenaunayova syndromu, u které byl zjištěn genetický základ.
Čtěte také:
Rizikové faktory
V přítomnosti vrozených srdečních vad existuje zvýšené riziko vzniku vrozeného aneurysmatu srdečního svalu a v přítomnosti vrozené hypertrofie dochází k vyboulení stěny levé komory. [ 1 ]
Aneuryzma hrudní aorty se s větší pravděpodobností vytvoří v případě vrozené vady bikuspidální aortální chlopně. Hrozba aneuryzmatu plicní chlopně je však mnohem vyšší, pokud je u novorozenců, zejména u předčasně narozených dětí, otevřené oválné okno. [ 2 ]
Intrakraniální aneuryzmata se tvoří častěji než jiná u dětí s vrozenou koarktací aorty, stejně jako s konstitučními vaskulopatiemi, zejména anomáliemi cév Willisova kruhu (z nichž arteriální hypoplazie je považována za nejčastější). [ 3 ], [ 4 ]
Téměř ve všech případech hrají klíčovou roli genetické rizikové faktory a přítomnost rodinné anamnézy aneurysmat, která naznačuje predispozici k jejich vzniku.
Patogeneze
Hlavní patogenezí aneuryzmat hlavních cév u Marfanova syndromu je mutace v genu FBN1 a porucha produkce proteinu extracelulární matrix cévní stěny fibrilinu-1. To mění strukturu a mechanické vlastnosti medie, středního obalu (tunica media) arteriálních cév. Kromě aneuryzmat vzestupného segmentu aorty mají pacienti s tímto syndromem insuficienci aortální chlopně.
Mechanismus vzniku aneurysmatu (včetně slezinové tepny) v důsledku vaskulárního typu Ehlers-Danlosova syndromu je vysvětlen určitými genovými mutacemi, které vedou k křehkosti kolagenu typu I a III - vláknitých proteinů mezibuněčné hmoty tkáně cévní stěny. Takové změny také oslabují cévní stěnu a působením hemodynamických sil průtoku krve v nejslabším místě nebo v oblasti zvýšeného "hemodynamického stresu" se postupně rozšiřuje a vybouluje.
Patogeneze arteriálních aneuryzmat u Loes-Dietzova syndromu je spojena s mutacemi v genech pro receptor transformujícího růstového faktoru (TGF-β). Odborníci se domnívají, že příčinou je fragmentace a ztráta elastinu, jehož vlákna zajišťují reverzibilní elasticitu stěn velkých cév. Poškození těchto vláken vede ke snížení kontraktility cévní stěny. Současně se molekuly proteoglykanů hromadí v mediální mezibuněčné matrici, což snižuje stabilitu kolagenu typu I, který udržuje pevnost cévních stěn.
Při mukopolysacharidóze dochází k hromadění glykosaminoglykanů, velkých lineárních polysacharidů (sacharidů), které narušují uspořádání kolagenních fibril, v endoteliálních a mediálních buňkách výstelky cévní stěny.
Kromě progresivní tvorby renálních cyst se předpokládá, že mutace PKD1/PKD2 u polycystické choroby ledvin způsobují defekty kolagenu a u pacientů s tímto dědičným onemocněním dochází k disekci aortální a cervikálně-hlavní tepny a tvorbě aneurysmatu - koronárních tepen, interatriálního septa a intrakraniálních cév.
Jak ukázaly histologické studie z posledních let, v případech neurofibromatózy typu 1 může být tvorba aneurysmatu břišní aorty nebo renálních tepen vedena proliferací Schwannových buněk periferního nervového systému, které se šíří do pochev cévní stěny.
U arteriálních cévních defektů mozku je mechanismus vzniku mozkového aneurysmatu spojen s poruchami v distribuci průtoku krve: karotické (tj. průtok krve ve vnitřních krčních tepnách - arteria carotis interna) a bazilární - v hlavní tepně zadní části mozku (arteria basilaris) a jejích větvích.
Symptomy vrozené aneurysma
Podle tvaru rozlišujeme aneurysmata vakovitá a vřetenovitá (fusiformní) a podle lokalizace se dělí na typy: mozkové (aneurysma mozkových cév), aneurysmata hrudní nebo břišní aorty, aneurysmata periferních tepen a další.
Vrozené aneuryzma aorty
Aneurysma postihující jakoukoli část aorty od aortální chlopně až po iliakální tepny. [ 5 ] Jejich příznaky jsou podrobně popsány v publikacích:
- Aneuryzma hrudní aorty
- Aneurysma vzestupné aorty.
- Aneuryzma břišní aorty
- Aneuryzmata aortálních větví
Vrozené aneurysma aortálního sinu
Aortální sinus nebo sinus Valsalva je jedno z anatomických rozšíření vzestupné aorty bezprostředně nad aortální chlopní. Aneuryzma aortálního sinu se nachází mezi prstencem aortální chlopně a oblastí vzestupné aorty, kde se konfigurace cévy opět stává tubulární (tzv. sinotubulární spojení). Takové aneuryzma vzniká v důsledku oslabení elastické laminy cévní stěny a tvoří se jako slepý divertikl. Nerupturované aneuryzma aortálního sinu je obvykle asymptomatické, ale pokud je dostatečně velké, může se projevit srdečními arytmiemi, aortální regurgitací, fibrilací síní a vést k akutnímu koronárnímu syndromu. [ 6 ], [ 7 ]
Vrozené aneurysma mozkových cév
Malé aneurysma nemusí způsobovat žádné příznaky, ale prvními příznaky zvětšení jsou náhlý nástup silných bolestí hlavy, nevolnosti a zvracení. Velké aneurysma může tlačit na mozkové struktury, což se projevuje rozšířením zornic a dvojitým viděním, poklesem víček, bolestí nad a za jedním okem, necitlivostí na jedné straně obličeje a záchvaty. [ 8 ], [ 9 ]
Více informací v materiálu - arteriální aneurysmata mozkových cév
Vrozené srdeční aneurysma
Všechny podrobnosti v publikaci - akutní a chronická srdeční aneuryzmata: ventrikulární, septální, postinfarktová, vrozená
Vrozené aneurysma síňového septa
Aneuryzma síňového septa je vrozená malformace síňového septa, lokalizovaná vakovitá deformace, která obvykle vzniká v oválné jamce a během srdečního cyklu často vyboulí do jedné nebo obou síní. [ 10 ]
Patologie může být spojena s vrozenými srdečními vadami, zejména s otevřeným oválným okénkem, prolapsem chlopně a otevřeným ductus arteriosus, ale ve třetině případů se projevuje jako izolovaná anomálie.
Mezi hlavní příznaky patří: dušnost, zejména při fyzické námaze; únava; otok dolních končetin a břicha; arytmie a palpitace; srdeční šelesty (při poslechu). [ 11 ]
Vrozené žilní aneurysma
Jedná se o vzácné malformace – cévní anomálie, které se mohou vyskytnout ve většině velkých žil; nejčastější lokalizací jsou dolní končetiny (více než 75 % případů), horní končetiny (až 10 % případů). Aneurysma vnitřní jugulární žíly (nejčastěji vřetenovitého tvaru) tvoří 13 % případů, přičemž dvě třetiny pacientů tvoří děti a dospívající. [ 12 ]
Aneurysma povrchové žíly se projevuje jako měkká podkožní masa (obvykle bezbolestná), která se zvětšuje při tlačení, kašlání nebo pláči. [ 13 ]
Komplikace a důsledky
Stěny aneurysmatu jsou patologicky změněné stěny cév, které ztratily své normální biomechanické vlastnosti a roztažnost jejich vybouleného místa je omezená, takže hlavními důsledky a komplikacemi jakéhokoli aneurysmatu jsou ruptura.
Všechny podrobnosti v publikaci - ruptura aneuryzmatu hrudní a břišní aorty
V tomto případě je ruptura vrozeného aneuryzmatu mozkových cév doprovázena subarachnoidálním krvácením, což má za následek dětskou úmrtnost více než 10 %.
Ruptura aneuryzmatu aortálního sinu je nebezpečná komplikace: ruptura pravého a nekoronárního sinu obvykle vede ke spojení mezi aortou a pravou síní nebo výtokovým traktem pravé komory, což má za následek výboj zleva doprava. To může způsobit přetížení pravé komory a pravostranné srdeční selhání.
Komplikací vrozeného aneuryzmatu srdce, zejména levé komory nebo síňového septa, může být zevní trombóza (v důsledku stáze krve); sraženina se může odtrhnout s potenciálem pro periferní arteriální embolii a kryptogenní cévní mozkovou příhodu. A když takové aneuryzma praskne, dochází k srdeční tamponádě.
Diagnostika vrozené aneurysma
Pro diagnostiku vrozeného aneurysmatu je nutné komplexní vyšetření, jehož základem je instrumentální diagnostika, která zahrnuje: EKG a echokardiografii; ultrazvuk a počítačovou tomografii hrudníku; ultrazvuk tepen vnitřních orgánů břišní dutiny; koronární angiografii a aortografii; kontrastní ventrikulografii; multispirální CT angiografii a MR angiografii; CT vyšetření lebeční báze; transkraniální dopplerografii atd.
Diferenciální diagnostika
Diferenciální diagnostika může být náročná. Například aneurysma ventrikulárního septa by mělo být odlišeno od velkých defektů septa, hypertrofie ventrikulární komory, Fallotova tetraedu a Eisenmengerova syndromu. A žilní aneurysma by mělo být odlišeno od křečových žil a trunkulárních žilních malformací, hemangiomu, laryngokély a enterické cysty.
Kdo kontaktovat?
Léčba vrozené aneurysma
Léčba obvykle začíná lékem, který pomáhá snižovat krevní tlak nebo uvolňovat cévy, aby se zabránilo prasknutí aneuryzmatu. Používají se také antikoagulancia – aby se zabránilo tvorbě krevních sraženin.
Číst:
U symptomatických vrozených aneuryzmat se chirurgická léčba provádí různými technikami (včetně endovaskulární) v závislosti na lokalizaci a velikosti aneuryzmatu. Více se dočtete v článku - operace arteriálních aneuryzmat
Prevence
Jako preventivní opatření, která jsou dnes k dispozici, odborníci uvádějí: lékařské a genetické poradenství, genetickou analýzu v těhotenství a také prenatální diagnostiku vrozených onemocnění.
Předpověď
Bohužel nemůže existovat příznivá prognóza pro všechny případy vrozených aneurysmat. Například, jak dlouho se dožívají pacienti s geneticky podmíněnými syndromy s patologií pojivové tkáně? Obecně je průměrná míra přežití u Marfanova syndromu asi 70 let, u Ehlers-Danlosova syndromu asi 50 let a u Loes-Dietzova syndromu něco málo přes 35 let. A ve většině případů dochází k úmrtí v důsledku ruptury aneurysmatu, krvácení do mozku nebo disekce hrudní/břišní aorty.
Pacienti s rupturovaným aneuryzmatem aortálního sinu obvykle umírají do jednoho roku od stanovení diagnózy – v důsledku městnavého srdečního selhání, uvádějí kliničtí lékaři.