Lékařský expert článku

Nové publikace
Neuroblastom u dětí: příčiny, diagnostika, léčba
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
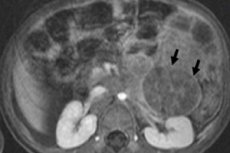
V dětské onkologii je jedním z nejčastějších extrakraniálních novotvarů u dětí neuroblastom, což je embryonální maligní nádor neuroblastů nervové lišty, tedy embryonálních (nezralých) nervových buněk sympatického nervového systému.
Epidemiologie
Podle statistik Mezinárodní rizikové skupiny pro neuroblastom (INRG) představuje neuroblastom přibližně 8 % všech onkologických onemocnění u dětí na celém světě a je na třetím místě v prevalenci po leukémii a mozkových nádorech.
Podle jiných údajů tvoří neuroblastom asi 28 % všech případů rakoviny u kojenců. Více než třetina případů neuroblastomu je diagnostikována u dětí mladších jednoho roku; průměrný věk diagnózy je 19–22 měsíců. Více než 90 % diagnostikovaných případů se vyskytuje u dětí ve věku od dvou do pěti let (s převahou chlapců); vrchol incidence je pozorován ve věku od dvou do tří let a případy u dětí starších pěti let tvoří méně než 10 %.
Příčiny neuroblastomy
Při studiu příčin neuroblastomu vědci dospěli k závěru, že tento nádor u dětí vzniká v důsledku sporadických genetických mutací během embryogeneze nebo raného postnatálního vývoje. Co však tyto genové změny způsobuje, není známo, protože nebyl zjištěn žádný vliv teratogenních faktorů prostředí.
Tyto nádory se mohou vyskytnout kdekoli, včetně mediastina, krku, břicha, nadledvin, ledvin, páteře a pánve.
Ve vzácných případech může být neuroblastom u kojenců spojen s dědičnou mutací. Zejména s mutací v genu membránového proteinu CD246 na chromozomu 2 - enzymu tyrosinkinázy ALK, který zajišťuje mezibuněčnou komunikaci a hraje důležitou roli ve fungování nervového systému; v genu proteinu PHOX2B (na chromozomu 4), který se podílí na zrání nervových buněk.
Neuroblastom může být také spojen s dětskou neurofibromatózou typu 1,Beckwithovým-Wiedemannovým syndromem a hyperinzulinemickou hypoglykémií (nesidioblastózní pankreatitidou).
Rizikové faktory
Dnes je dědičnost uznávána jako rizikový faktor pro rozvoj neuroblastomu u dětí - přítomnost tohoto nádoru v rodinné anamnéze, stejně jako vrozené anomálie spojené s genovými mutacemi během nitroděložního vývoje. To platí zejména pro případy vývoje několika novotvarů v různých orgánech.
Žádný z exogenních faktorů, které zvyšují riziko tohoto nádoru, nebyl vědci identifikován.
Patogeneze
Mechanismus vývoje neuroblastomů je způsoben poruchami diferenciace a zrání buněk neurální lišty – bilaterálních buněčných linií, které se tvoří na okrajích neurální trubice z ektodermální zárodečné vrstvy lidského embrya. Tyto buňky migrují (pohybují se) a diferencují se do mnoha typů buněk: senzorických a autonomních neuronů, neuroendokrinních buněk a buněk dřeně nadledvin, buněk kraniofaciální chrupavky a kostí a také pigmentových buněk.
U neuroblastomu migrované neuroblasty nedozrávají, ale pokračují v růstu a dělení, čímž tvoří nádor. Patogeneze jeho vzniku je spojena s následujícími genovými mutacemi:
- s duplikací části chromozomální sekvence nebo duplikací segmentů genu LMO1 na chromozomu 11, kódujícího protein RBTN1 v buňkách nervové lišty embrya;
- se změnou počtu kopií genu NBPF10 na chromozomu 1q21.1, kódujícího protein DUF1220, který řídí proliferaci lidských nervových kmenových buněk. Tyto poruchy vedou buď k duplikaci tohoto chromozomu, nebo k jeho deleci – absenci části DNA;
- se změnami v genu pro supresor tumoru ATRX (na chromozomu Xq21.1);
- s přítomností dalších kopií (amplifikací) genu transkripčního faktoru N-Myc na chromozomu 2, který kóduje jeden z transkripčních faktorů (protein vázající DNA), jenž reguluje aktivitu ostatních genů a řídí proliferaci prekurzorových buněk během tvorby proteinů pro tvorbu tkání a orgánů plodu. Amplifikace tohoto genu jej mění v onkogen, což vyvolává narušení buněčného cyklu, zvýšenou proliferaci buněk a tvorbu nádorů.
Symptomy neuroblastomy
První příznaky neuroblastomu jsou nespecifické a mohou zahrnovat ztrátu chuti k jídlu (a úbytek hmotnosti), únavu při krmení, horečku a bolest kloubů.
Klinické příznaky závisí na lokalizaci primárního nádoru a přítomnosti metastáz (které se vyskytují v 60–73 % případů).
Primární neuroblastom je velmi často lokalizován v dřeni nadledvin, která má podobný původ jako nervové buňky. U dětí mladších jednoho roku je neuroblastom nadledvin diagnostikován v 35–40 % případů. Mezi jeho příznaky patří bolesti břicha, horečka, úbytek hmotnosti, bolesti kostí, anémie nebo souběžný Pepperův syndrom: difúzní poškození jater s těžkou hepatomegalií a syndromem respirační tísně.
Retroperitoneální neuroblastom neboli retroperitoneální neuroblastom u dětí s růstem začíná tlačit na močový měchýř nebo střeva, což může způsobit problémy s močením nebo defekací, otoky nohou (u chlapců otéká šourek).
Neuroblastom mediastina u dětí (mediastinální neuroblastom) často tlačí na horní dutou žílu, což může způsobit otok obličeje, krku, paží a horní části hrudníku (kůže se zbarví do modročervena s podkožními uzlíky). Objevuje se kašel a sípání, dýchací potíže (dušnost) nebo polykání (dysfagie); zvětšené lymfatické uzliny jsou zaznamenány na krku, nad klíční kostí a v podpaží.
Šíření nádorových buněk do kostní dřeně vede k anémii, trombocytopenii a leukopenii se sklonem ke krvácení.
A s metastázami v periorbitální oblasti se kolem očí objevují tmavé kruhy nebo modřiny. Takový nádor může také způsobovat bolesti hlavy a závratě, exoftalmii (vyboulení očních bulv) a v důsledku stlačení nervových zakončení - pokles očních víček (ptóza) a zmenšení velikosti zornic (mióza).
Abdominální neuroblastom nebo neuroblastom břišní dutiny u dětí vede k tvorbě hmatatelných těsnění v břiše, jeho nadýmání, ztrátě chuti k jídlu, zácpě a zvýšenému krevnímu tlaku. Nádor tlačící na míchu nebo nervový kořen může vést k necitlivosti a slabosti končetin, neschopnosti stát, plazit se nebo chodit. Pokud jsou postiženy kosti, může se objevit bolest kostí.
V případě nádoru 3.-4. stupně v břišní dutině s poškozením lymfatických uzlin mohou nádorové buňky proniknout do parenchymu ledvin a následně se u dětí vyvine rozsáhlý neuroblastom ledviny, což vede k narušení jejích funkcí.
Etapy
- Neuroblastom 1. stádia je primární nádor, který je lokalizován a izolovaný v jedné oblasti těla; lymfatické uzliny na obou stranách nejsou postiženy.
- Neuroblastom ve stádiu 2. Ve stádiu 2A je primární nádor omezen na jednu oblast, ale je velký; nejsou postiženy bilaterální lymfatické uzliny. Ve stádiu 2B jsou lymfatické uzliny na straně těla, kde se nádor nachází, pozitivní na metastázy.
- Neuroblastom 3. stádia: primární nádor prochází míchou nebo středovou linií těla, v lymfatických uzlinách se nacházejí jednostranné nebo oboustranné metastázy.
- Neuroblastom stádium 4: nádor se rozšířil do vzdálených lymfatických uzlin, kostní dřeně, kostí, jater nebo jiných orgánů. Stádium 4S se stanoví u dětí mladších jednoho roku s lokalizovaným primárním nádorem s diseminací do kůže, jater nebo kostní dřeně.
Mezinárodní systém pro stanovení stadia rizika neuroblastomu (INRGSS)
INRGSS používá rizikové faktory definované zobrazovacími metodami (IDRF), což jsou faktory pozorované při zobrazovacích vyšetřeních, které mohou znamenat, že odstranění nádoru bude obtížnější.
INRGSS rozděluje neuroblastomy do 4 stádií:
- L1: Nádor se nerozšířil z místa, kde vznikl, a neprorostl do životně důležitých struktur. Je omezen na jednu část těla, například na krk, hrudník nebo břicho.
- L2: Nádor se nerozšířil (nemetastázoval) daleko od místa, kde začal (například mohl růst z levé strany břicha na levou stranu hrudníku), ale má alespoň jeden IDRF.
- M: Nádor metastázoval do vzdálené části těla (s výjimkou nádorů ve stádiu RS).
- RS: Metastatické onemocnění u dětí mladších 18 měsíců, u kterého se rakovina rozšířila pouze do kůže, jater a/nebo kostní dřeně.
Komplikace a důsledky
Neuroblastom se vyznačuje komplikacemi a následky, jako jsou:
- šíření (metastázy) do lymfatických uzlin, kostní dřeně, jater, kůže a kostí;
- komprese míchy (která může způsobit bolest a vést k paralýze);
- rozvoj paraneoplastického syndromu (v důsledku působení určitých chemických látek vylučovaných nádorem, stejně jako antigenu disialogangliozidu GD2 exprimovaného jeho buňkami), který se projevuje rychlými mimovolními pohyby očí, zhoršenou koordinací, svalovými křečemi a průjmem;
- relapsy po ukončení primární terapie (jak ukazuje klinická praxe, vysoce rizikové neuroblastomy mají relaps v 50 % případů).
Diagnostika neuroblastomy
Diagnóza podezření na neuroblastom u dítěte vyžaduje vyšetření, laboratorní testy a zobrazovací metody.
Provádějí se krevní a močové testy na katecholaminy (norepinefrin a dopamin) a kyseliny homovanilové nebo vanilylmandlové (vznikající při metabolismu těchto hormonů); krevní test na neurospecifickou enolázu, enzymově vázaný imunosorbentní test (ELISA) krevního séra a analýza kostní dřeně (jejíž vzorek se odebírá aspirační punkcí). Provádí se test DNA k určení mutací a biopsie k cytomorfologickému vyšetření nádorové tkáně.
Po odebrání vzorků biopsie jsou tyto vzorky odeslány do laboratoře, kde je pod mikroskopem vyšetří patolog (lékař se speciálním výcvikem v identifikaci rakovinných buněk). Na vzorcích se také často provádějí speciální laboratorní testy, které prokáží, zda se jedná o neuroblastom.
Pokud se jedná o neuroblastom, laboratorní testy mohou také pomoci určit, jak rychle může nádor růst nebo se šířit, a také jaké léčebné postupy by mohly fungovat nejlépe.
Instrumentální diagnostika vizualizuje novotvar pomocí ultrazvuku, rentgenu, MRI nebo CT, PET se zavedením 18F-fluorodeoxyglukózy nebo MIBG skenování - scintigrafie s metajodbenzylguanidinem. [ 1 ]
Diferenciální diagnostika
Diferenciální diagnóza zahrnuje benigní ganglioneurom, ganglioneuroblastom, rabdomyosarkom a nefroblastom.
Kdo kontaktovat?
Léčba neuroblastomy
U neuroblastomu závisí léčba na rizikové skupině pacienta (stadiu nádorového procesu), lokalizaci nádoru, genomických vlastnostech nádorových buněk a věku dítěte. Může zahrnovat monitorování, chirurgický zákrok, chemoterapii, radioterapii, imunoterapii a transplantaci hematopoetických kmenových buněk.
Neoadjuvantní nebo adjuvantní (předoperační nebo pooperační) chemoterapie neuroblastomu u dětí, stejně jako jakákoli chemoterapie rakoviny, se podává v cyklech: lék se podává několik dní po sobě, po kterých následuje přestávka, aby se tělo zotavilo. Cykly se obvykle opakují každé tři až čtyři týdny.
Používají se následující léky (a jejich kombinace): cyklofosfamid, cisplatina nebo karboplatina, doxorubicin (adriamycin), vinkristin, etoposid.
Mezi časté nežádoucí účinky chemoterapie patří vypadávání vlasů, ztráta chuti k jídlu, únava, nevolnost a zvracení, afty v ústech, průjem nebo zácpa. Chemoterapie může poškodit kostní dřeň a způsobit snížení počtu krvinek.
Cílená imunoterapie (zaměřená na nádorový antigen GD2) využívá léky ze skupiny monoklonálních protilátek (anti-GD2 MAb) Dinutuximab (Unituxin) a Naxitamab. Podávají se intravenózně prodlouženou infuzí v kombinaci s faktorem stimulujícím kolonie granulocytů a makrofágů (cytokin GM-CSF) a interleukinem-2.
Mezi nežádoucí účinky těchto léků patří bolest (často velmi silná), snížený krevní tlak, zvýšená srdeční frekvence, dušnost (s možným otokem dýchacích cest), zvýšená teplota, nevolnost, zvracení a průjem, změny buněčného a minerálního složení krve.
Aby se snížilo riziko recidivy rakoviny po vysokodávkové chemoterapii a transplantaci kmenových buněk, jsou děti s vysoce rizikovým neuroblastomem léčeny systémovými retinoidy, kyselinou 13-cis-retinovou (isotretinoinem). [ 2 ]
Chirurgická léčba neuroblastomu – odstranění nádoru, například otevřená adrenalektomie nebo laparoskopická resekce neuroblastomu nadledvin; lymfektomie (odstranění postižených lymfatických uzlin) atd. [ 3 ]
U vysoce rizikového neuroblastomu lze použít radioterapii.[4 ]
Prevence
Vzhledem k příčinám neuroblastomu u dětí může být jediným preventivním opatřením genetické poradenství při plánování těhotenství. Je však třeba mít na paměti, že tento nádor je spojen s dědičnými mutacemi pouze v 1-2 % případů.
Předpověď
Infantilní neuroblastom má schopnost spontánní regrese.
Prognostické markery
- Vysoce rizikové nádory, stejně jako neuroblastom u dětí všech věkových skupin a všech stadií (kromě stadia 4S) – se zvýšenou expresí genu N-MYC a amplifikací onkogenu N-Myc – mají nepříznivou prognózu, která ovlivňuje délku života.
- Nádorové buňky, kterým chybí určité části chromozomů 1 nebo 11 (známé jako delece 1p nebo 11q), mají horší prognózu. S horší prognózou je také spojena přítomnost části chromozomu 17 navíc (zvýšení chromozomu 17q).
- Neuroblastomové buňky s velkým množstvím DNA mají lepší prognózu, zejména u dětí mladších 2 let.
- Neuroblastomy, které mají více neurotrofinových receptorů, zejména receptoru nervového růstového faktoru TrkA, mají lepší prognózu.
Přežití podle rizikové skupiny dětské onkologické skupiny (COG)
- Skupina s nízkým rizikem: Děti v skupině s nízkým rizikem mají 5letou míru přežití vyšší než 95 %.
- Středně riziková skupina: Děti ve střední rizikové skupině mají 5letou míru přežití 90 % až 95 %.
- Vysoce riziková skupina: Děti ve vysoce rizikové skupině mají 5letou míru přežití přibližně 50 %.
Přibližně 15 % úmrtí na rakovinu v dětství je způsobeno neuroblastomem. Šance na dlouhodobé přežití u tohoto vysoce rizikového maligního onemocnění nepřesahuje 40 %. Celková pětiletá míra přežití je 67–74 %, 43 % ve věkové skupině od jednoho do čtyř let a více než 80 % u neuroblastomu diagnostikovaného v prvním roce života.
Использованная литература