
Cystická fibróza
Naposledy posuzováno: 23.04.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Cystická fibróza - genetická autozomálně recesivní monogenní onemocnění charakterizované zhoršenou sekreci žláz s vnější sekrecí životně důležité orgány s lézemi především dýchacího a trávicího systému, závažné a nepříznivou prognózou.
 [1]
[1]
Epidemiologie
Incidence cystické fibrózy se pohybuje od 1: 2500 do 1: 4,600 novorozenců. Každoročně se na světě rodí asi 45 000 pacientů s cystickou fibrózou. Frekvence přenašečů cystické fibrózy 3-4%, po celém světě, existuje asi 275 milionů lidí - .. Nositeli genu, z nichž asi 5 milionů žije v Rusku, o 12,5 milionů -. V ch zemí.
Příčiny cystická fibróza
Cystická fibróza se přenáší autosomálně recesivním typem. Gen cystické fibrózy se nachází v 7 autosomech, obsahuje 27 exonů a skládá se z 250 000 párů nukleotidů.
V jednom genu je možné mnoho mutací, z nichž každá je charakteristická pro určitou populaci nebo geografickou oblast. Je popsáno více než 520 mutací, z nichž nejběžnější je delta-P-508, tj. Nahrazení aminokyseliny fenylalaninu v 508 polohách.
Patogeneze
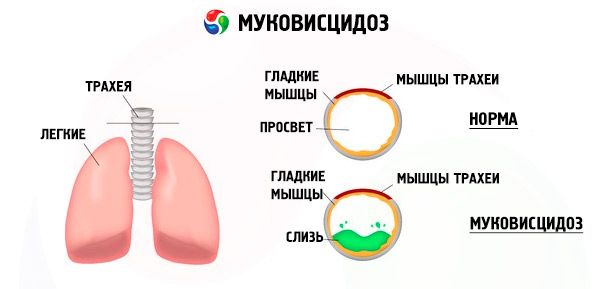
Vzhledem k mutací genu cystické fibrózy je narušena struktura a funkce proteinu, nazývaného transmembránový regulátor CFTR-cystická fibróza. Tento protein působí jako chloridový kanál zapojený do metabolismu vody a elektrolytů epiteliálních buněk bronchopulmonálního systému, gastrointestinálního traktu, pankreatu, jater a reprodukčního systému. V důsledku porušení funkce a struktury proteinu CFTR se ionty chloru hromadí uvnitř buňky. Cl - ). To vede ke změně elektrického potenciálu v lumen výstupním kanálem, čímž se vstupem ve velkém množství iontů sodíku (Na + ), z lumen do toku buněk a dále zvyšuje absorpci vody z pericelulárních prostoru.
Z důvodu těchto změn zahušťuje nejtajnější exokrinní žlázy, jeho evakuace je přerušeno, což vede k výrazným sekundární poruchy v orgánech a systémech, nejvýraznější v bronchopulmonálních a trávicího ústrojí.
Bronchi vyvinout chronické zánětlivé proces různé intenzity, což výrazně narušena funkce řasinkový epitel, hlen se stává velmi viskózní, hustá, velmi obtížné být evakuován, je možno pozorovat, stagnace vytvořen bronhiolo- a bronchiektázie, která se stává více a více převládající v průběhu času. Tyto změny vedou ke zvýšení hypoxie a vzniku chronického plicního srdce.
Pacienti s cystickou fibrózou jsou extrémně náchylní k vývoji chronického zánětlivého procesu v bronchopulmonálním systému. Je to způsobeno výraznými poruchami v systému lokální bronchopulmonární ochrany (snížení hladiny IgA, interferonu, fagocytární funkce alveolárních makrofágů a leukocytů).
Důležitou roli ve vývoji chronického zánětu v bronchopulmonálním systému patří alveolární makrofágy. Vyrábějí velké množství IL-8, což dramaticky zvyšuje chemotaxi neutrofilů v bronchiálním stromu. Neutrofily se hromadí ve velkých množstvích v průduškách a spolu s epiteliální buňky vylučují množství provovospalitelnyh cytokinů, včetně IL-1, 8, 6, faktor nekrotizující nádory, a leukotrieny.
Důležitou roli v patogenezi lézí bronchopulmonálního systému hraje také vysoká aktivita enzymu elastáza. Existuje exogenní a endogenní elastáza. První je produkována bakteriální flórou (zejména Pseudomonas aeruginosa), druhou neutrofilními leukocyty. Elastáza zničí epitel a další strukturální prvky průdušek, což přispívá k dalšímu narušení mukociliární transportní a nejrychlejší tvorbě bronchiektáz.
Neutrofilní leukocyty také vylučují jiné proteolytické enzymy. Působí proti vlivu proteolytických enzymů, a tím chrání bronchopulmonární systém z jejich poškození vlivem a1-anppripsin a leykoproteaz inhibitor sekreční. Nicméně, bohužel, u pacientů s cystickou fibrózou jsou tyto ochranné faktory potlačeny významným množstvím neutrofilní proteázy.
Všechny tyto okolnosti přispívají k zanesení infekce do bronchopulmonální systému, vývoj chronické hnisavého zánětu průdušek. Kromě toho je třeba poznamenat, že defektní protein kódovaný genem pro cystickou fibrózu, změní funkční stav bronchiálního epitelu, který podporuje adhezi bakterií na bronchiálního epitelu, zejména Pseudomonas aeruginosa.
Spolu s patologií bronchopulmonálního systému při cystické fibróze dochází také k výrazné porážce pankreatu, žaludku, tlustého a tenkého střeva a jater.
Symptomy cystická fibróza
Cystická fibróza se projevuje různými klinickými příznaky. U novorozenců se onemocnění může projevit meconial ileus. Vzhledem k nedostatku, nebo dokonce úplné nepřítomnosti trypsinu mekonia stává velmi husté, viskózní, že se hromadí v ileocekální oblasti. Dále rozvíjet ileus, což se projevuje s intenzivní zvracení žluči, nadýmání, nedostatkem příznaků meconium zánět pobřišnice, rychlým růstem klinických projevů syndromu těžké intoxikace. Dítě může zemřít v prvních dnech života, pokud nedojde k okamžitému chirurgickému zákroku.
V méně závažných případech je charakteristickým znakem cystické fibrózy hojná, častá stolice, mast, s velkým množstvím tuku, s velmi nepříjemným zápachem. U 1/3 pacientů dochází k prolapsu konečníku.
Následkem toho přetrvává střevní dysfunkce u pacientů, dochází k rozvoji malabsorpčního syndromu, vážnému narušení fyzického vývoje, závažné hypovitaminóze.
V prvním druhém roce života dítěte jsou příznaky bronchopulmonální systému (mírná forma onemocnění), která se projevuje kašel, který může být velmi závažné a připomínají kašle na černý kašel. Kašel doprovázený cyanózou, dušnost, oddělení hustého sputa, původně sliznice a pak hnisavý. Postupně se vytváří klinický obraz chronické obstrukční bronchitidy a bronchiektázy, emfyzému plic a respiračního selhání. Děti jsou extrémně náchylné k akutním respiračně-virovým a bakteriálním infekcím, které přispívají k exacerbacím a progresi bronchopulmonální patologie. Možný vývoj infekčně závislého bronchiálního astmatu.
U dětí ve školním věku se cystická fibróza může projevit jako "střevní kolika". Pacienti se stěžují na závažné paroxysmální bolesti v břiše, nadýmání, opakované zvracení. Při hmatání břicha jsou identifikovány husté útvary, které se nacházejí v projekci tlustého střeva - výkaly, které jsou smíšené s hustým hustým hlenem. Děti jsou velmi náchylné k rozvoji hypochloremické alkalózy kvůli nadměrnému odstranění solí potu v horkém počasí, zatímco na pokožce dítěte se objevuje "sůl mráz".
Porážka bronchopulmonálního systému u dospělých
Porážka bronchopulmonární systému u pacientů s cystickou fibrózou (plicní forma onemocnění) je charakterizována vývojem chronický hnisavý obstrukční bronchitida, bronchiektázie, chronická pneumonie, rozedma plic, respirační selhání, plicní srdce. Někteří pacienti vyvinuli pneumotorax a jiné komplikace cystické fibrózy: atelektázy, plicní absces, hemoptýzou, plicní krvácení, infekce závislá na průduškového astmatu.
Pacienti se stěžují na bolestivý paroxysmický kašel s velmi viskózním, těžko oddělitelným mukopurulentním sputem, někdy s příměsí krve. Dyspnoe je navíc spíše charakteristické při fyzickém namáhání a poté v klidu. Dyspnoe je způsobeno průduškou. Mnoho pacientů si stěžuje na chronickou rinitidu způsobenou polypózou a sinusitidou. Existuje také významná slabost, progresivní pokles výkonnosti, časté akutní respiračně-virové onemocnění. Při vyšetření se věnuje pozornost bledosti pokožky, otok tváře, cyanóza viditelných sliznic, výrazná dušnost. Při vývoji dekompenzovaného plicního srdce se objeví edém na nohou. Mohlo by dojít ke ztluštění terminálových falangů prstů rukou v podobě tympanických tyčinek a nehtů - ve formě přesných hodin. Hrudník získává tvar hlaveň (v důsledku rozvoje emfyzému).
Při perkuse plic se určují příznaky emfyzému - zvuk boxu, ostré omezení pohyblivosti plicní hrany, snížení spodního okraje plic. Při auskultaci plic se objevuje tvrdé dýchání s prodlouženým výdechem, rozptýlené suché sipky, vlhké médium a malé bublinky. Při výrazném emfyzému plic je dýchání ostře oslabeno.
Extratorakální projevy cystické fibrózy
Extrapulmonární projevy cystické fibrózy mohou být poměrně výrazné a často se vyskytují.
Porážka pankreatu
Nedostatečnost exokrinní funkce pankreatu různé závažnosti je pozorována u 85% pacientů s cystickou fibrózou. S mírným lézí pankreatu špatného trávení a malabsorpční syndromy nepřítomné, existují pouze laboratorní projevy exokrinní insuficience (nízké hladiny trypsinu a lipázy v krvi a dvanáctníku, často vyjádřena steatorrhea). Je známo, že k zabránění syndromu poruchy poruchy se vylučuje pouze 1 až 2% celkové lipázy. Klinicky se projevují pouze významná porušení vnější sekreční funkce.
Za normálních podmínek se sekrece tekuté konzistence bohaté na enzymy produkuje v acini pankreatu. S průběhem sekrece přes vylučovací kanál je obohacen vodou a anionty a stává se ještě více tekutým. U cystické fibrózy v důsledku narušení struktury a funkce regulátoru transmembránové (chloridové kanály) v pankreatu tajemství neobdrží dostatečné kapalina se stává viskózní, a rychlost jeho posuvu ductless dramaticky zpomaluje. Tajné proteiny jsou uloženy na stěnách malých vylučovacích kanálů, v důsledku kterých se jejich obstrukce vyvíjí. Jak postupovala nemoc nakonec vyvine ničení a atrofii acinus - tvořil chronické pankreatitidy s exokrinní pankreatické nedostatečnosti. To se klinicky odráží ve vývoji syndromů maldigestie a malabsorpce. Pankreatická nedostatečnost je hlavní příčinou tukové malabsorpce u cystické fibrózy, ale obvykle se pozoruje při výrazně exprimován nedostatkem lipázy. Forsher a Durie (1991) ukazují, že v nepřítomnosti pankreatické lipázy a absorbován tuk se odštěpí o 50-60% v důsledku přítomnosti žaludeční a slin (sublingvální), lipázové aktivity, která je v blízkosti spodní hranici normálu. Kromě porušení trávení a vstřebávání tuku je porušení rozdělení a opětovné absorpce proteinů. S výkaly se ztratí asi 50% bílkovin, které jsou krmeny potravinami. Absorpce sacharidů trpí v menší míře i přes nedostatek alfa-amylázy, ale může být podstatně narušen metabolismus sacharidů.
Porážka pankreatu je vyjádřena ve vývoji syndromu maldigestie a malabsorpce s významnou ztrátou hmotnosti, bohatou tukovou stolicí.
Vývoj Poruchy trávení a syndromy malabsorpce také podporuje těžkou dysfunkci střevní žlázy, poruchy uvolňování a sníženou střevní šťávy obsažené střevní enzymy.
Syndromy maldigestie a malabsorpce jsou také nazývány střevní formou cystické fibrózy.
Porušení přírůstkové funkce pankreatu (diabetes mellitus) je pozorováno u pacientů s cystickou fibrózou v pozdních stádiích onemocnění (u 2% dětí a u 15% dospělých)
Léze jater a žlučovodů
U 13% pacientů se smíšenou a střevní cystickou fibrózou se rozvíjí cirhóza. Nejběžnější je u mutací W128X, delta-P508 a X1303K. U 5-10% pacientů se objevila biliární cirhóza s portální hypertenzí. Podle Welcha, Smitha (1995) se u 86% pacientů s cystickou fibrózou vyskytují klinické, morfologické, laboratorní a instrumentální příznaky poškození jater.
Mnoho pacientů s cystickou fibrózou také rozvíjí chronickou cholecystitidu, často kalkulovanou.
Porušení funkce sexuálních žláz
U pacientů s cystickou fibrózou se může vyskytnout azoospermie, která je příčinou neplodnosti. Snížení plodnosti je také charakteristické pro ženy.
Etapy
Existují tři stupně závažnosti plicní formy cystické fibrózy.
Mírná forma cystické fibrózy je charakterizována vzácnými exacerbacemi (ne častěji než jednou za rok), během období remisí prakticky chybí klinické projevy, nemocní jsou schopni pracovat.
Průběh mírné závažnosti - exacerbace je pozorován 2-3krát ročně a trvá asi 2 měsíce a déle. V akutní fázi je pozorován intenzivní kašel s těžko oddělitelným sputum, dyspnoe dokonce i bez zanedbatelné fyzické námahy, tělesné teploty v subfabrilu, obecné slabosti, pocení. Současně dochází k porušení exokrinní funkce pankreatu. Ve fázi remisí není pracovní kapacita zcela obnovena, dyspnoe zůstává během fyzické námahy.
Silný průběh je charakterizován velmi častými exacerbacemi nemoci. Neexistují prakticky žádné remisie. V klinickém obrazu je těžké respirační selhání, symptomologie chronického plicního srdce, často dekompenzovaná, charakterizovaná hemoptýzou, v popředí. Existuje významný pokles tělesné hmotnosti, pacienti jsou zcela zdravotně postižení. Zpravidla je těžká bronchopulmonální patologie doprovázena výrazným poškozením funkce pankreatu.
Formuláře
- Broncho-plicní léze
- Opakovaná a rekurentní pneumonie s prodlouženým průběhem.
- Abscesní pneumonie, zejména u kojenců.
- Chronická pneumonie, zejména bilaterální.
- Bronchiální astma, refrakterní na konvenční terapii.
- Rekurentní bronchitida, bronchiolitida, zejména při naočkování Pseudomonas aeruginosa.
- Změny v gastrointestinálním traktu
- Meconial ileus a jeho ekvivalenty.
- Syndrom zhoršené intestinální absorpce neznámého původu.
- Obstrukční žloutenka u novorozenců s prodlouženým průběhem.
- Cirhóza jater.
- Diabetes mellitus.
- Gastroezofageální reflux.
- Holelitiaz.
- Prolaps konečníku.
- Změny v jiných orgánech a systémech
- Poruchy růstu a vývoje.
- Zpožděný sexuální vývoj.
- Mužská neplodnost.
- Polypy nosu.
- Sibs z rodin s pacienty s cystickou fibrózou.
[24]
Komplikace a důsledky
Komplikace z gastrointestinálního traktu zahrnují:
- Diabetes mellitus se rozvíjí u 8-12% pacientů starších 25 let.
- Fibrotická kolonopatie.
- Mekonium ileus v novorozeneckém období (12% dětí s CF, distální střevní obstrukce syndrom, rektální výhřez, peptického vředu a refluxní choroby jícnu.
Komplikace z jater:
- Mastná degenerace jater (u 30-60% pacientů),
- Ohnisková biliární cirhóza, mnohoinodulární biliární cirhóza a související portální hypertenze.
Portální hypertenze někdy vede k smrti v důsledku křečových žil jícnu.
Prevalence cholecystitidy a cholelitiázy je vyšší u pacientů s cystickou fibrózou než u jiných jedinců.
Zpožděná puberta a snížená plodnost a další komplikace. Většina mužů má azoospermii a nedostatečné rozvinutí vas deferens.
Diagnostika cystická fibróza
Obvyklý krevní test je charakterizován anémií s různou závažností, obvykle normálně nebo hypochromně. Anémie má mnohočetnou genezi (snížení absorpce železa a vitamínu B12 ve střevě v důsledku výskytu malabsorpčního syndromu). Možná leukopenie, s vývojem hnisavé bronchitidy a pneumonie - leukocytóza, zvýšení ESR.
Obecná analýza moči - bez významných změn, ve vzácných případech je bezvýznamná proteinurie.
Koprologické vyšetření - existuje steatorrhoea, creatorrhea. Becker (1987) doporučuje měření ve výkalích chymotrypsinu a mastných kyselin. Před stanovením chymotrypsinu v stolici je nutné zrušit příjem trávicích enzymů nejméně 3 dny před testem. Při cystické fibróze se sníží množství chymotrypsinu v stolici a počet mastných kyselin se zvýší (normální uvolňování mastných kyselin je méně než 20 mmol / den). Je třeba vzít v úvahu, že zvýšené vylučování mastnými kyselinami ve stolici je také pozorováno, pokud:
- Nedostatek konjugovaných mastných kyselin v tenkém střevě v selhání jater, obstrukce žlučovodu, významný bakteriální kolonizace tenkého střeva (to je doprovázeno intenzivním hydrolýzou žlučových kyselin);
- niles;
- celiakie (s rozvojem malabsorpčního syndromu);
- enteritida;
- střevní lymfomy;
- Whippleova choroba;
- potravinové alergie;
- zrychlený průchod jídelních hmot za průjem různého původu, karcinoidní syndrom, tyreotoxikózu.
Biochemická analýza krve - snížení celkových proteinů a hladiny albuminu zvýšení alfa-2 a gama-globulin, bilirubin a transaminázy (s onemocněním jater), snížená aktivita amylázy, lipázy, trypsinu a hladiny železa vápníku (na vývoj syndromu špatného trávení, malabsorpční).
Analýza sputa - přítomnost velkého počtu neutrofilních leukocytů a mikroorganismů (s rozsypem sputa).
Vyšetření absorpční funkce tenkého střeva a exokrinní funkce pankreatu - zjištěné významné porušení.
Rentgenové vyšetření plic - odhaluje změny, jejichž závažnost závisí na závažnosti a fázi onemocnění. Nejcharakterističtější změny jsou:
- zvýšení intenzity plicního vzorku v důsledku peribronchiálních změn intersticií;
- rozšíření kořenů plic;
- vzor lobulární, subsegmentální nebo dokonce segmentální atelectázy plic;
- zvýšená průhlednost plicních polí, zejména v horních částech, nízká stálost a nedostatečná pohyblivost membrány, rozšíření vaginálního prostoru (projev emfyzému plic);
- segmentální nebo polysegmentární infiltrace plicní tkáně (s vývojem pneumonie).
Bronchography - detekovat změny způsobené ce obstrukční bronchiální viskózní sputum (bronchiální fragmentace náplň kontrast, nerovnostem průdušky rozbití jev, k výraznému snížení počtu bočních větví) a bronhoekgazy (válcovité, smíšené), lokalizované zejména v nižších částí plic.
Bronchoskopie - detekuje difúzní purulentní bronchitidu s hojným množstvím hustého, viskózního sputa a fibrinózních filmů.
Spirography - i v časných stádiích onemocnění odhaluje obstrukční respirační typ poruchy (snížení FVC, FEV1 index Tiffno), omezující (snížen VC) nebo častěji obstrukční-omezující (snížení vitální kapacita, FVC, FEV1, Tiffno index).
Test potu (studium potních elektrolytů) Gibsonem a Cookem - stimulace pocení pomocí elektroforézy s pilokarpinem a následné stanovení v chloridu potu. Doerehuk (1987) popisuje vzorek následujícím způsobem. Elektroforéza pilokarpinu se vytváří v oblasti předloktí, elektrický proud je 3 mA. Po vyčištění pokožky destilovanou vodou se pot sbírá filtrační papír aplikovaný na stimulovanou oblast, pokrytý gázou, aby nedošlo k jejímu odpaření. Po 30-60 minutách se filtrační papír odstraní a eluuje se v destilované vodě. Změřte množství odebraného potu. Pro získání spolehlivých výsledků je nutné shromáždit nejméně 50 mg (s výhodou 100 mg) potu.
Při koncentraci chloridu vyšší než 60 mmol / l je diagnóza cystické fibrózy považována za pravděpodobnou; při koncentraci chloridu vyšší než 100 mmol / l - spolehlivé; zatímco rozdíl v koncentraci chloru a sodíku by neměl přesáhnout 8-10 mmol / l. Hadson (1983) doporučuje vzorek s prednisolonem při hraniční hodnotě obsahu sodíku a chloridu (užívající 5 mg perorálně po dobu 2 dnů, následované elektrolyty v potu). U osob, které nemají cystickou fibrózu, se hladina sodíku v potu sníží na hodnotu spodní hranice normy s cystickou fibrózou - nemění se. Vzorek pocení se doporučuje každému dítěti s chronickým kašlem.
Analýza krevních skvrn nebo vzorků DNA u hlavních mutací genu cystické fibrózy je nejcitlivějším a specifickým diagnostickým testem. Tato metoda je však vhodná pro země, kde je frekvence mutace delta-P508 vyšší než 80%. Navíc je technika velmi drahá a technicky složitá.
Prenatální diagnóza cystické fibrózy se provádí stanovením isoenzymů alkalické fosfatázy v plodové vodě. Tato metoda je možná s 18-20 týdny těhotenství.
Hlavní kritéria pro diagnostiku cystické fibrózy jsou následující:
- indikace v historii dětské retardace ve fyzickém vývoji, recidivující chronické nemoci dýchacího ústrojí, dyspeptické poruchy a průjmy, přítomnost cystické fibrózy v nejbližší rodině;
- chronická obstrukční bronchitida, často recidivující, s vývojem bronchiektázie a emfyzému, často recidivující pneumonií;
- chronická recidivující pankreatitida s výrazným snížením exokrinní funkce, syndromem malabsorpce;
- zvýšený obsah chlóru v potu pacienta;
- neplodnost se zachovalou sexuální funkcí.
Úspěšnou diagnózu a diferenciální diagnostiku cystické fibrózy usnadňuje identifikace rizikových skupin.
Program vyšetření cystické fibrózy
- Obecná analýza krve, moči, sputa.
- Bakteriologická analýza sputa.
- Koprologická analýza.
- Biochemická analýza krve: Stanovení celkového proteinu a proteinových frakcí, glukózy, bilirubinu, transamináz, alkalické fosfatázy, gamaglutamyltransferáza, draslík, vápník, železo, lipázy, amylázy, trypsinu.
- Vyšetření exokrinní funkce pankreatu a funkce střevní absorpce.
- Rentgenová a plicní radiografie, CT plic.
- ECG.
- Echokardiografie.
- Bronchoskopie a bronchografie.
- Spirografie.
- Test potu.
- Konzultace genetika.
- Analýza krevních skvrn nebo vzorků DNA pro hlavní mutace genu cystické fibrózy.
Jaké testy jsou potřeba?
Kdo kontaktovat?
Léčba cystická fibróza
Typ a závažnost příznaků cystické fibrózy mohou být velmi odlišné, takže neexistuje typický léčebný plán, je individuální v každém případě.
Terapie se skládá z následujících léčebných opatření:
- Dýchací cvičení a posturální odvodnění pomáhají zbavit se hustého hlenu, který se hromadí v plicích. Některé metody čištění dýchacích cest vyžadují pomoc od členů rodiny, přátel nebo pulmonologa. Mnoho lidí používá nafukovací vestu, která vibruje s vysokou frekvencí.

- Inhalační léky, které působí bronchodilatanci, drenáž (mukolytika) a antibakteriální účinky (např. Fluorochinolony).
- Přípravky obsahující pankreatické enzymy pro zlepšení trávení. Tyto léky jsou užívány s jídlem.
- Multivitamíny (včetně vitamínů rozpustných v tucích).
V roce 2015 FDA schválila druhou drogu pro léčbu cystické fibrózy, která postihuje defektní protein známý jako CFTR. První lék, tzv. Modulátor CFTR, byl schválen v roce 2012. Očekává se, že modulátory CFTR mohou prodloužit životnost některých lidí s cystickou fibrózou po desítky let.
Chirurgická léčba může být zapotřebí k léčbě následujících respiračních komplikací:
- Pneumotorax, masivní recidivující nebo přetrvávající hemoptýza, nosní polypy, perzistentní a chronická sinusitida.
- Meconiální obstrukce, intususcepce, prolaps konečníku.
Transplantace plic se provádí v terminálním stádiu onemocnění.
Předpověď
Průměrný věk přežití pacientů s cystickou fibrózou se pohybuje od 35 do 40 let. Průměrný věk přežití u mužů je vyšší než u žen.
Díky moderním léčebným postupům dosahuje dospělost 80% pacientů. Nicméně cystická fibróza významně omezuje funkční schopnosti pacienta. Lék na tuto chorobu dosud nebyl vyvinut.