Lékařský expert článku

Nové publikace
Priony - původci prionových onemocnění
Naposledy posuzováno: 06.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Pomalé virové infekce se vyznačují zvláštními kritérii:
- neobvykle dlouhá inkubační doba (měsíce, roky);
- specifická léze orgánů a tkání, především centrálního nervového systému;
- pomalý, stálý postup onemocnění;
- nevyhnutelným smrtelným následkem.

Některé patogeny, které způsobují akutní virové infekce, mohou také způsobovat pomalé virové infekce. Například virus spalniček někdy způsobuje SSPE a virus zarděnek způsobuje progresivní vrozenou zarděnku a zarděnkovou panencefalitidu.
Typickou pomalou virovou infekci u zvířat způsobuje virus visna/madi, což je retrovirus. Je původcem pomalé virové infekce a progresivního zápalu plic u ovcí. Bílá hmota mozková je zničena, rozvíjí se paralýza (visna - chřadnutí); dochází k chronickému zánětu plic a sleziny.
Nemoci, které se svými znaky podobají pomalým virovým infekcím, jsou způsobeny priony – původci prionových infekcí. Prionová onemocnění jsou skupinou progresivních poruch centrálního nervového systému lidí a zvířat. U lidí je narušena funkce centrálního nervového systému, dochází ke změnám osobnosti a poruchám pohybu. Příznaky onemocnění obvykle trvají několik měsíců až několik let a končí smrtí. Dříve byly prionové infekce považovány za součást tzv. původců pomalých virových infekcí.
Některé agens, které způsobují prionová onemocnění, se hromadí nejprve v lymfoidních tkáních. Priony se po vstupu do mozku hromadí ve velkém množství a způsobují amyloidózu (extracelulární dysproteinózu, charakterizovanou ukládáním amyloidu s rozvojem atrofie a sklerózy tkáně) a astrocytózu (proliferace astrocytárních neuroglií, hyperprodukce gliových vláken). Vznikají fibrily, agregáty proteinů nebo amyloidu a spongiformní změny v mozku (přenosné spongiformní encefalopatie). V důsledku toho se mění chování, zhoršuje se koordinace pohybů, rozvíjí se vyčerpání s fatálním koncem. Nevytváří se imunita. Prionová onemocnění jsou konformační onemocnění, která se vyvíjejí v důsledku nesprávného skládání (porušení správné konformace) buněčných proteinů nezbytných pro normální fungování těla. Cesty přenosu prionů jsou různé:
- alimentární cestou - infikované produkty živočišného původu, potravinářské přídatné látky ze syrových hovězích orgánů atd.:
- přenos krevní transfuzí, podáváním léků živočišného původu, transplantací orgánů a tkání, použitím infikovaných chirurgických a zubních nástrojů;
- přenos imunobiologickými přípravky (je známa infekce 1500 ovcí PrP''' vakcínou z mozkového formalinu od nemocných ovcí).
Patologické priony se po vstupu do střeva transportují do krve a lymfy. Po periferní replikaci ve slezině, slepém střevě, mandlích a dalších lymfoidních tkáních se periferními nervy dostávají do mozku (neuroinvaze). Přímý průnik prionů do mozku přes hematoencefalickou bariéru je možný. Dříve se věřilo, že centrální nervový systém je jedinou tkání, ve které se patologické priony hromadí, ale objevily se studie, které tuto hypotézu změnily. Ukázalo se, že akumulace prionů ve slezině je spojena se zvýšením počtu a fungováním folikulárních dendritických buněk.
 [
[ Vlastnosti prionů
Normální buněčná izoforma prionového proteinu s molekulovou hmotností 33-35 kDa je určena genem pro prionový protein (prionový gen - PrNP se nachází na 20. lidském chromozomu). Normální gen se nachází na povrchu buňky (ukotvený v membráně glykoproteinem molekuly), citlivý na proteázu. Reguluje přenos nervových impulsů, denní cykly, oxidační procesy, podílí se na metabolismu mědi v centrálním nervovém systému a na regulaci dělení kmenových buněk kostní dřeně. Kromě toho se prionový gen nachází ve slezině, lymfatických uzlinách, kůži, gastrointestinálním traktu a folikulárních dendritických buňkách.
Proliferace patologických prionů
K transformaci prionů do pozměněných forem dochází, když je narušena kineticky řízená rovnováha mezi nimi. Proces je urychlen zvýšením množství patologického (PrP) nebo exogenního prionu. PrP je normální protein ukotvený v buněčné membráně. PrP' je globulární hydrofobní protein, který na povrchu buňky tvoří agregáty sám se sebou a s PrP'': v důsledku toho se PrP' transformuje na PrP'' a cyklus pokračuje. Patologická forma PrP''' se hromadí v neuronech, což dává buňce houbovitý vzhled.
Kuru
Prionová choroba, dříve běžná mezi Papuany (ve smyslu třes nebo chvění) ve východní části ostrova Nová Guinea. Infekční vlastnosti nemoci prokázal K. Gajdusek. Původce onemocnění se přenáší potravou v důsledku rituálního kanibalismu - konzumace nedostatečně tepelně upraveného, priony infikovaného mozku zemřelých příbuzných. V důsledku poškození centrální nervové soustavy dochází k narušení pohybu a chůze, objevuje se zimnice a euforie („smějící se smrt“). Inkubační doba trvá 5-30 let. Nemocný umírá po roce.
Creutzfeldt-Jakobova choroba
Prionová choroba, která se projevuje demencí, poruchami zraku a mozečku a poruchami pohybu s fatálním koncem po 4-5 měsících onemocnění u klasické varianty Creutzfeldt-Jakobovy choroby a po (3-14 měsících) u nové varianty Creutzfeldt-Jakobovy choroby. Inkubační doba může dosáhnout 20 let. Jsou možné různé cesty infekce a příčiny onemocnění:
- při konzumaci nedostatečně tepelně ošetřených živočišných produktů, jako je maso a mozky krav s bovinní spongiformní encefalopatií;
- během transplantace tkání, jako je transplantace rohovky, krevní transfuze, použití hormonů a jiných biologicky aktivních látek živočišného původu, použití katgutu, kontaminovaných nebo nedostatečně sterilizovaných chirurgických nástrojů, prosektorálních manipulací;
- v případě hyperprodukce PrR a dalších stavů, které stimulují proces přeměny PrR' na PrR".
Onemocnění se může také vyvinout v důsledku mutace nebo inzerce v oblasti prionového genu. Familiární povaha onemocnění je běžná kvůli genetické predispozici k Creutzfeldtově-Jakobově chorobě. U nové varianty Creutzfeldt-Jakobovy choroby se poruchy rozvíjejí v mladším věku (průměrný věk 28 let), na rozdíl od klasické varianty (průměrný věk 65 let). U nové varianty Creutzfeldt-Jakobovy choroby se abnormální prionový protein hromadí nejen v centrálním nervovém systému, ale také v lymforetikulárních tkáních, včetně mandlí.
Gerstmann-Sträussler-Scheinkerův syndrom
Dědičné prionové onemocnění, doprovázené demencí, hypotonií, poruchami polykání (dysfagií), dysartrií. Často má familiární charakter. Inkubační doba je 5 až 30 let. Onemocnění se objevuje ve věku 50–60 let, jeho délka trvání se pohybuje od 5 do 13 let.
Dědičná fatální nespavost
Autoimunitní onemocnění s progresivní nespavostí, sympatickou hyperreaktivitou (hypertenze, hypertermie, hyperhidróza, tachykardie), tremorem, ataxií, multiklonálním selháním a halucinacemi. Spánek je závažně narušen. S progresí kardiovaskulárního selhání nastává smrt.
Škrábanec
Scrapie (z anglického scrape - škrábat) je prionové onemocnění ovcí a koz (svrab), které probíhá s poškozením centrálního nervového systému, progresivními poruchami hybnosti, silným svěděním kůže (svrab) a končí úhynem zvířete.
Bovinní spongiformní encefalopatie
Onemocnění skotu charakterizované poškozením centrálního nervového systému, zhoršenou koordinací pohybů a nevyhnutelnou smrtí zvířete. Epidemie onemocnění poprvé vypukla ve Velké Británii. Byla spojena s krmením zvířat masokostní moučkou obsahující patologické priony. Inkubační doba se pohybuje od 1,5 do 15 let. Mozek, mícha a oční bulvy zvířat jsou nejvíce infikovány.
Laboratorní diagnostika prionových onemocnění
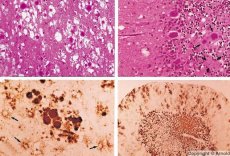
Během diagnostiky se zaznamenávají spongiformní změny v mozku, astrocytóza (glióza) a absence zánětlivých infiltrátů. Mozek se barví na amyloid. V mozkomíšním moku se detekují proteinové markery prionových poruch mozku (pomocí ELISA). Provádí se genetická analýza prionového genu (PCR).
Prevence prionových onemocnění
Pro dekontaminaci nástrojů a předmětů z prostředí se doporučuje autoklávování (při 134 °C po dobu 18 minut; při 121 °C po dobu 1 hodiny), spalování, dodatečné ošetření bělidlem a roztokem NaCl o koncentraci 1 % po dobu 1 hodiny. Pro nespecifickou profylaxi byla zavedena omezení používání léčivých přípravků živočišného původu a je zakázána produkce hypofyzárních hormonů živočišného původu. Transplantace tvrdé pleny mozkové je omezena. Při práci s dialýzami pacientů se používají gumové rukavice.