Lékařský expert článku

Nové publikace
Nemoc Charcot-Marie-Tooth.
Naposledy posuzováno: 12.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
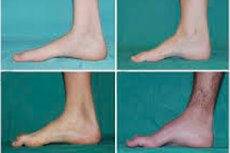
Peroneální svalová atrofie, Charcot-Marie-Toothův syndrom nebo onemocnění, je skupina chronických dědičných onemocnění s poškozením periferních nervů.
Podle MKN-10 je v sekci o onemocněních nervové soustavy kódem pro toto onemocnění G60.0 (hereditární motorická a senzorická neuropatie). Je také zařazeno do seznamu vzácných onemocnění.
Epidemiologie
Podle klinických statistik je prevalence všech typů Charcot-Marie-Toothova syndromu na 100 tisíc obyvatel 19 případů (podle jiných zdrojů jeden případ na 2,5–10 tisíc obyvatel).
CMT typu 1 představuje přibližně dvě třetiny případů (jeden případ na 5 000 až 7 000 obyvatel) a téměř 70 % z nich je spojeno s duplikací genu PMP22. Tímto typem onemocnění trpí více než 1,2 milionu lidí na celém světě.
Výskyt CMT typu 4 se odhaduje na 1–5 případů na 10 000 dětí. [ 1 ]
Příčiny Nemoc Charcot-Marie-Tooth
Podle klasifikace polyneuropatických syndromů se peroneální (fibulární) svalová atrofie, Charcot-Marie-Toothova neurální amyotrofie nebo Charcot-Marie-Toothova choroba (zkráceně CMT) vztahuje ke geneticky podmíněným motoricko-senzorickým polyneuropatiím. [ 2 ]
To znamená, že příčinami jeho výskytu jsou genetické mutace. A v závislosti na povaze genetických odchylek se rozlišují hlavní typy nebo druhy tohoto syndromu: demyelinizační a axonální. První skupina zahrnuje Charcot-Marie-Toothův syndrom typu 1 (CMT1), který vzniká v důsledku duplikace genu PMP22 na chromozomu 17, který kóduje transmembránový periferní myelinový protein 22. V důsledku toho dochází k segmentální demyelinaci axonální pochvy (výběžků nervových buněk) a ke snížení rychlosti vedení nervového signálu. Kromě toho mohou být mutace v některých dalších genech.
Axonální formou je Charcot-Marie-Toothova choroba typu 2 (CMT2), která postihuje samotné axony a je spojena s patologickými změnami v genu MFN2 v lokusu 1p36.22, kódujícím membránový protein mitofusin-2, který je nezbytný pro fúzi mitochondrií a tvorbu funkčních mitochondriálních sítí uvnitř periferních nervových buněk. Existuje více než tucet podtypů CMT2 (s mutacemi ve specifických genech).
Je třeba poznamenat, že v současné době bylo identifikováno více než sto genů, jejichž poškození, přenášené dědičností, způsobuje různé podtypy Charcot-Marie-Toothovy choroby. Například mutace v genu RAB7 vedou k CMT typu 2B; změna genu SH3TC2 (kódujícího jeden z membránových proteinů Schwannových buněk) způsobuje CMT typu 4C, která se projevuje v dětství a je charakterizována demyelinací motorických a senzorických neuronů (existuje tucet a půl forem typu 4 této choroby).
Vzácná CMT typu 3 (nazývaná Dejerine-Sottasův syndrom) se začíná rozvíjet v raném dětství a je způsobena mutacemi v genech PMP22, MPZ, EGR2 a dalších.
Pokud se CMT typu 5 objeví ve věku 5-12 let, pozoruje se nejen motorická neuropatie (ve formě spastické paraparézy dolních končetin), ale také poškození zrakového a sluchového nervu.
Svalová slabost a optická atrofie (se ztrátou zraku), stejně jako problémy s rovnováhou, jsou typické pro CMT typu 6. A u Charcot-Marie-Toothovy choroby typu 7 se nejen vyskytuje motoricko-senzorická neuropatie, ale také onemocnění sítnice ve formě retinitis pigmentosa.
U mužů je častější výskyt X-vázané CMT neboli Charcot-Marie-Toothovy choroby s tetraparézou končetin (slabost pohybu paží i nohou). Jedná se o demyelinizační typ a předpokládá se, že je důsledkem mutace v genu GJB1 na dlouhém raménku chromozomu X, který kóduje konexin 32, transmembránový protein Schwannových buněk a oligodendrocytů, který reguluje přenos nervových signálů. [ 3 ]
Rizikové faktory
Hlavním rizikovým faktorem pro CMT je rodinná anamnéza onemocnění, tedy u blízkých příbuzných.
Podle genetiků, pokud jsou oba rodiče nositeli autozomálně recesivního genu pro Charcot-Marie-Toothovu chorobu, je riziko narození dítěte, u kterého se toto onemocnění rozvine, 25 %. A riziko, že dítě bude nositelem tohoto genu (ale nebude mít žádné příznaky), se odhaduje na 50 %.
V případě dědičnosti vázané na chromozom X (kdy se mutovaný gen nachází na chromozomu X ženy) existuje 50% riziko, že matka předá gen svému synovi, u kterého se rozvine CMT. Onemocnění se nemusí projevit při narození dívky, ale synové (vnoučata) dcery mohou zdědit vadný gen a onemocnění se rozvine.
Patogeneze
U jakéhokoli typu Charcot-Marie-Toothovy choroby je její patogeneze způsobena dědičnou anomálií periferních nervů: motorických (pohybových) a senzorických (citlivých).
Pokud je typ CMT demyelinizační, pak destrukce nebo defekt myelinové pochvy, která chrání axony periferních nervů, vede ke zpomalení přenosu nervových impulsů v periferním nervovém systému – mezi mozkem, svaly a smyslovými orgány.
U axonálního typu onemocnění jsou postiženy samotné axony, což negativně ovlivňuje sílu nervových signálů, která je nedostatečná pro plnou stimulaci svalů a smyslových orgánů.
Přečtěte si také:
Jak se přenáší Charcot-Marie-Toothův syndrom? Vadné geny se mohou dědit autozomálně dominantně nebo autozomálně recesivně.
Nejběžnější typ, autozomálně dominantní dědičnost, nastává, když existuje jedna kopie mutovaného genu (kterou nosí jeden z rodičů). Pravděpodobnost přenosu CMT na každé narozené dítě se odhaduje na 50 %. [ 4 ]
Při autozomálně recesivní dědičnosti jsou k rozvoji onemocnění potřebné dvě kopie defektního genu (jedna od každého rodiče, který nevykazuje žádné známky onemocnění).
Ve 40–50 % případů se vyskytuje autozomálně dominantní dědičná demyelinizace, tj. CMT typ 1; ve 12–26 % případů axonální CMT, tj. typ 2. A v 10–15 % případů je pozorována dědičnost vázaná na chromozom X. [ 5 ]
Symptomy Nemoc Charcot-Marie-Tooth
První příznaky tohoto onemocnění se obvykle objevují v dětství a dospívání a postupně se rozvíjejí po celý život, ačkoli se syndrom může projevit i později. Kombinace symptomů je variabilní a rychlost progrese onemocnění, stejně jako jeho závažnost, nelze předvídat.
Mezi typické příznaky počátečního stádia patří zvýšená celková únava; snížený tonus (slabost) svalů chodidel, kotníků a holení; nedostatek reflexů. To ztěžuje pohyb nohou a vede k dysbazii (poruše chůze) v podobě vyššího zvedání nohou, často s častými zakopnutími a pády. Mezi příznaky Charcot-Marie-Toothovy choroby u malého dítěte může patřit výrazná nemotornost a pro věk necharakteristické potíže s chůzí spojené s bilaterálním poklesem chodidla. Charakteristické jsou také deformity chodidel: vysoká klenba (prohnutá noha) nebo silné ploché nohy, zakřivené (kladívkovité) prsty.
V případě chůze po špičkách na pozadí svalové hypotonie může neurolog mít podezření, že dítě má CMT typu 4, kdy děti nemusí být schopny chodit v adolescenci.
S postupující nemocí se svalová atrofie a slabost šíří do horních končetin, což ztěžuje jemnou motoriku a běžné úkony s rukama. Snížené hmatové vjemy a schopnost cítit teplo a chlad, stejně jako necitlivost nohou a rukou, naznačují poškození axonů senzorických nervů.
U Charcot-Marie-Toothovy choroby typu 3 a 6, která se projevuje v dětství, se pozoruje senzorická ataxie (porucha koordinace pohybů a rovnováhy), svalové záškuby a třes, poškození lícního nervu, atrofie zrakového nervu s nystagmem a ztráta sluchu.
V pozdějších stádiích se může objevit nekontrolovatelný třes (tremor) a časté svalové křeče; problémy s pohybem mohou vést k rozvoji bolesti: svalové, kloubní, neuropatické.
Komplikace a důsledky
Charcot-Marie-Toothova choroba může mít komplikace a následky, jako například:
- častější výrony a zlomeniny;
- kontraktury spojené se zkrácením periartikulárních svalů a šlach;
- skolióza (zakřivení páteře);
- dýchací potíže – když jsou poškozena nervová vlákna, která inervují svaly bránice:
- ztráta schopnosti samostatného pohybu.
Diagnostika Nemoc Charcot-Marie-Tooth
Diagnóza zahrnuje klinické vyšetření, anamnézu (včetně rodinné anamnézy), neurologické a systémové vyšetření.
Provádějí se testy k ověření rozsahu pohybu, citlivosti a šlachových reflexů. Nervové vedení lze posoudit pomocí instrumentální diagnostiky – elektromyografie nebo elektroneuromyografie. Může být také nutný ultrazvuk nebo magnetická rezonance. [ 6 ]
Genetické nebo DNA testování k detekci nejčastějších genetických mutací, které způsobují CMT ve vzorku krve, je omezené, protože DNA testy v současné době nejsou k dispozici pro všechny typy onemocnění. Více informací naleznete v části Genetické testování.
V některých případech se provádí biopsie periferního nervu (obvykle surálního nervu).
Diferenciální diagnostika
Diferenciální diagnóza zahrnuje další periferní neuropatie, Duchennovu svalovou dystrofii, myelopatické a myastenické syndromy, diabetickou neuropatii, myelopatie u roztroušené a amyotrofické laterální sklerózy, Guillain-Barréův syndrom, trauma peroneálního nervu a jeho atrofii (včetně skřípnutí mezi bederními ploténkami páteře), poškození mozečku nebo thalamu, jakož i vedlejší účinky chemoterapie (během léčby cytostatiky, jako je vinkristin nebo paklitaxel). [ 7 ]
Kdo kontaktovat?
Léčba Nemoc Charcot-Marie-Tooth
Léčba tohoto dědičného onemocnění dnes zahrnuje cvičební terapii (zaměřenou na posílení a protažení svalů); ergoterapii (která pomáhá pacientům se svalovou slabostí v rukou); a používání ortopedických pomůcek k usnadnění chůze. V případě potřeby se předepisují léky proti bolesti nebo antikonvulziva. [ 8 ]
V případech těžké ploché nohy lze provést osteotomii a v případě deformace paty je indikována chirurgická korekce – artrodéza. [ 9 ]
Probíhá výzkum jak genetické složky onemocnění, tak i metod jeho léčby. Použití kmenových buněk, některých hormonů, lecitinu nebo kyseliny askorbové zatím nepřineslo pozitivní výsledky.
Ale díky nedávnému výzkumu se v léčbě Charcot-Marie-Toothovy choroby v blízké budoucnosti skutečně může objevit něco nového. Francouzská společnost Pharnext tak od roku 2014 vyvíjí a od poloviny roku 2019 probíhají klinické studie léku PXT3003 pro léčbu CMT typu 1 u dospělých, který potlačuje zvýšenou expresi genu PMP22, zlepšuje myelinaci periferních nervů a zmírňuje neuromuskulární příznaky.
Specialisté z lékařské společnosti Sarepta Therapeutics (USA) pracují na vytvoření genové terapie pro Charcot-Marie-Toothovu chorobu typu 1. Tato terapie bude využívat neškodný adeno-asociovaný virus (AAV) rodu Dependovirus s lineárním jednovláknovým DNA genomem, který do těla přenese gen NTF3 kódující protein neurotrofin-3 (NT-3) nezbytný pro fungování Schwannových nervových buněk.
Společnost Helixmith zahájí do konce roku 2020 klinické studie genové terapie Engensis (VM202) vyvinuté v Jižní Koreji k léčbě svalových symptomů u karpometakarpální myelody typu 1. [ 10 ]
Prevence
Prevencí CMT může být genetické poradenství budoucích rodičů, zejména pokud má někdo z páru rodinnou anamnézu tohoto onemocnění. Byly však identifikovány i případy de novo bodových genových mutací, tedy bez přítomnosti onemocnění v rodinné anamnéze.
Během těhotenství lze pravděpodobnost Charcot-Marie-Toothovy choroby u budoucího dítěte zkontrolovat biopsií choriových klků (od 10. do 13. týdne těhotenství) a také analýzou plodové vody (v 15.-18. týdnu).
Předpověď
Obecně platí, že prognóza různých typů Charcot-Marie-Toothovy choroby závisí na klinické závažnosti, ale ve všech případech onemocnění postupuje pomalu. Mnoho pacientů má postižení, i když to nezkracuje délku života.