
Dědičná nefritida (Alportův syndrom) u dětí
Naposledy posuzováno: 23.04.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Hereditární nefritida (Alportova syndromu) - geneticky podmíněné neimunitní dědičné glomerulopatie vykazující hematurie (někdy proteinurie), progresivní snížení funkce ledvin u chronického selhání ledvin vývoji je často spojován s senzorineurální hluchotou a zrakově postižené.
Poprvé byla nemoc popsána v roce 1902 LGGuthrie, která pozorovala rodinu v několika generacích, z níž byla pozorována hematurie. V roce 1915 členové téže rodiny AFHurst popsali vývoj uremie. V roce 1927 A Alport nejprve identifikoval hluchotu u několika příbuzných s hematurií. V padesátých letech minulého století bylo v takovém onemocnění popsáno poškození očí. V roce 1972 u pacientů s dědičnou hematurií morfologicky vyšetřujícím ledvinnou tkáň, Hinglais et al. Odhalila nerovnoměrnou expanzi a delaminaci glomerulárních bazálních membrán. V roce 1985 byla identifikována genetická základna dědičné nefritidy - mutace v genu kolagenu typu IV (Fiengold et al., 1985).
Zkoumání genetické povahy onemocnění umožnilo dospět k závěru, že rozdíly v fenotypových projevech dědičné nefritidy (se ztrátou sluchu nebo bez ní) jsou způsobeny stupněm exprese mutantního genu. V současné době jsou všechny klinické varianty považovány za projevy jedné choroby a termín "dědičná nefritida" je synonymem pro výraz "Alportův syndrom".
Podle epidemiologických studií se dědičná nefritida vyskytuje s frekvencí 17 na 100 000 dětí.
Příčiny Alportova syndromu
Genetickým základem onemocnění je mutace genu a-5 kolagenového řetězce typu IV. Tento typ univerzální pro bazálních ledvin membrány, kochleární zařízení, kapsle čočky, sítnice a rohovky, které je ukázáno na studiích s použitím monoklonálních protilátek proti kolagenu frakci. V poslední době naznačují možnost použití DNA sondy pro prenatální diagnostiku dědičných nefritid.
Je zdůrazněn význam testování všech členů rodiny pomocí DNA sondy k identifikaci nosičů mutantního genu, což je velmi důležité pro provádění lékařského genetického poradenství rodin s tímto onemocněním. Nicméně až 20% rodin nemá příbuzné s onemocněním ledvin, což naznačuje vysoký výskyt spontánních mutací v abnormálním genu. Většina pacientů s dědičnou nefritidou v rodinách má jedince s onemocněním ledvin, ztrátou sluchu a patologií zraku; které mají jednoho nebo více předků, protože sňatek příbuzných osob zvyšuje pravděpodobnost získání stejných genů od obou rodičů. Autozomálně dominantní a autozomálně recesivní a dominantní, spojená s chromozómem X přenosové cesty jsou zavedena.
Děti rozlišují tři varianty dědičné nefritidy: Alportův syndrom, dědičná nefritida bez ztráty sluchu a rodinná benigní hematurie.
Alportův syndrom - dědičná nefritida se poškozením sluchu. Základem je kombinovaný defekt ve struktuře kolagenu bazální membrány glomerulů ledvin, struktur ucha a oka. Gén klasického Alportova syndromu se nachází v místě 21-22 q dlouhého ramena chromozomu X. Ve většině případů se dědí dominantním typem spojeným s X chromozómem. V tomto ohledu je u mužů Alportův syndrom obtížnější, protože u žen je funkce mutantního genu kompenzována zdravou alelou druhého intaktního chromozomu.
Genetická základna vývoje dědičné nefritidy jsou mutace genů alfa řetězců kolagenu typu IV. To je známé jako šesti-řetězce kolagenu typu IV G: A5 a A6 geny řetězců (Sol4A5 a Sol4A5) jsou umístěny na dlouhém rameni chromozomu X v 21-22q zóně; geny řetězců a3 a a4 (Co4A3 a Co4A4) - na 2. Chromozómu; geny a1- a a2-řetězců (Co4A1 a Co4A2) - na 13. Chromozómu.
Ve většině případů (80-85%) je X-spojený typ dědičnosti onemocnění spojen s poškozením genu Co4A5 v důsledku delece, bodových mutací nebo poruch spojování. V současnosti je nalezeno více než 200 mutací genu Kol4A5, které jsou odpovědné za narušení syntézy a5 řetězců kolagenu typu IV. V tomto druhu dědictví se onemocnění projevuje u dětí obou pohlaví, ale u chlapců je to obtížnější.
Mutace v lokusích genů Co4A3 a Co4A4, které jsou zodpovědné za syntézu řetězců a3 a a4 řetězce kolagenu typu IV, se dědí autosomálně. Podle výzkumu je autozomálně dominantní typ dědičnosti pozorován u 16% případů dědičné nefritidy, autosomálně recesivní - u 6% pacientů. Existuje přibližně 10 mutací genů Co4A3 a Co4A4.
Výsledkem mutací je narušení procesů shromažďování kolagenu typu IV, což vede k narušení jeho struktury. Kolagen typu IV je jednou z hlavních složek glomerulární bazální membrány, kochleárního aparátu a čočky oka, jejíž patologie bude odhalena na klinice dědičné nefritidy.
Kolagen typu IV, část glomerulární bazální membráně, sestává v podstatě ze dvou řetězců a1 (IV) a a2 jednoho řetězce (IV), a také obsahuje A3, A4, A5 řetězce. Nejčastěji, když je X-vázaná dědičnost Sol4A5 mutace doprovázen nedostatek A3, A4, A5 a A6 řetězce kolagenu typu IV, ve struktuře, a počet O1 a A2 řetězců do glomerulárních zvyšuje bazální membrány. Mechanismus tohoto jevu je nejasné, předpokládá se, že důvodem je, že po transkripci mRNA změny.
Nedostatek A3, A4 a A5 řetězce na struktury typu IV kolagenu bazální membráně glomerulů výsledků v ztenčení a křehkosti časných stádiích Alportova syndromu, který se sám projevuje klinicky většinu hematurie (někdy hematurie nebo proteinurie pouze proteinurii), ztráty a lenticonus sluchu. Další postup onemocnění vede k zahuštění, a narušení bazální membrány propustnosti v pozdních stádiích onemocnění, s růstem těchto kolagenu typu V a VI, které se projevují ve zvýšení proteinurie a snížené funkci ledvin.
Povaha mutace podléhající dědičné nefritidě velmi určuje její fenotypový projev. Když X chromozomu, delecí se simultánním mutace a Sol4A6 Sol4A5 genů zodpovědných za syntézu A5 a A6 řetězce kolagenu typu IV, v kombinaci s Alportova syndromu leiomyomatosis jícnu a pohlavních orgánů. Podle studií s mutacemi genu Sol4A5 spojených s delecí jsou označeny velké vážnosti patologického procesu, kombinace s renálním poškozením extrarenální projevy a raný vývoj chronického selhání ledvin, ve srovnání stochechnoy mutace tohoto genu.
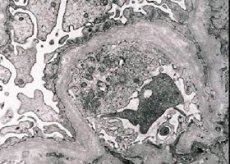
Morfologicky, elektronová mikroskopie odhaluje ztenčení a delaminaci glomerulárních bazálních membrán (zejména lamina densa) a přítomnost elektronově hustých granulí. Léze glomerulu může být u stejného pacienta nerovnoměrné, od minimální fokální léze mesangia až po glomerulosklerózu. Glomerulitida u Alportova syndromu je vždy imunologicky negativní, což ji odlišuje od glomerulonefritidy. Charakteristické jsou vývoj atrofie kanálu, infiltrace lymfohistiocytů, přítomnost "pěnových buněk" s inkorporací lipidů - lipofágů. S progresí onemocnění se zjistí zesílení a výrazná destrukce bazálních glomerulových membrán.
Jsou odhaleny určité změny ve stavu imunitního systému. U pacientů s dědičnou nefritidou je zaznamenáno snížení hladiny IgA a tendence ke zvýšení koncentrace IgM v krvi, úroveň IgG může být zvýšena v časných stádiích vývoje onemocnění a později se snižovat. Možná zvýšení koncentrace IgM a G je druh kompenzační odpovědi v reakci na deficit IgA.
Funkční aktivita systému T-lymfocytů je snížena; Je označen selektivní redukce B-lymfocytů, které je odpovědné za syntézu Ig, rozdělených fagocytární imunitu odkaz, zejména v důsledku zhroucení procesů chemotaxe a intracelulární trávení neutrofilů
Ve studii s renální biopsie u pacientů s Alportovým syndromem elektronovým mikroskopem, ultrastrukturální změny pozorovány glomerulární bazální membrány: ztenčení a Štěpení narušení glomerulární bazální membrány se změnou v jeho tloušťce a nerovnostem. V časných stádiích dědičné nefritidy defekt určuje ztenčení a křehkost glomerulární bazální membrány.
Ředění glomerulárních membrán je příznivějším znakem a je častější u dívek. Konstantnější elektronové mikroskopické vlastnosti v dědičné nefritidě je štěpení bazální membrány a závažnost její destrukce koreluje se závažností procesu.

Symptomy Alportova syndromu u dětí
První příznaky Alportova syndromu ve formě izolovaného syndromu moče jsou častěji zjištěny u dětí prvních tří let života. Ve většině případů je nemoc zjištěna náhodou. Močový syndrom se objevuje během preventivního vyšetření dítěte před vstupem do dětské instituce nebo během ARVI. V případě výskytu patologie v moči během ARVI. V dědičné nefritidě, na rozdíl od získané glomerulonefritidy, neexistuje latentní doba.
V počátečním stádiu onemocnění trpí dítě málo, charakteristickým znakem je přetrvávání a přetrvávání močového syndromu. Jedním z hlavních příznaků je hematurie různého stupně, pozorovaná ve 100% případů. Zvýšení stupně hematurie je zaznamenáno během nebo po infekcích dýchacího traktu, při fyzické námaze nebo po preventivních očkováních. Proteinurie ve většině případů nepřesahuje 1 g / den, na počátku onemocnění může být nestabilní, protože postup vede k růstu proteinurie. Pravidelně močový sediment může mít leukocyturiu s převahou lymfocytů, což je spojeno s vývojem intersticiálních změn.
Později dochází k porušení částečných funkcí ledvin, zhoršení celkového stavu pacienta: intoxikace, svalová slabost, arteriální hypotenze, často postižení sluchu (zejména u chlapců), někdy i zhoršení vidění. Intoxikace se projevuje jako bledost, únava, bolesti hlavy. V počáteční fázi onemocnění je ztráta sluchu ve většině případů zjištěna pouze audiografií. Úbytek sluchu u Alportova syndromu se může vyskytnout v různých dětských věkových obdobích, ale nejčastěji je ztráta sluchu diagnostikována ve věku 6-10 let. Ztráta sluchu u dětí začíná na vysokých frekvencích, dosahuje významného stupně vzdušného a kosočtvercového průchodu, od zvukovodu až po ztrátu sluchu. Úbytek sluchu může být jedním z prvních příznaků onemocnění a může předcházet močový syndrom.
Ve 20% případů mají pacienti s Alportovým syndromem změny v očích. Nejběžnější anomálie z čočky: spherofokiya, lentikonus přední, zadní nebo smíšené, různé katarakty. U rodin s Alportovým syndromem je významný výskyt myopie. Řada badatelů jsou neustále v těchto rodin slavit dvoustranné perimakulyarnye změny jako světlé bělavé nebo žlutavé granulací ve žlutém tělísku. Považují tento symptom za konstantní symptom, který má vysokou diagnostickou hodnotu u Alportova syndromu. C. S. Chugh a kol. (1993) pro oftalmologické studie ukázala, pacienti Alportova syndromu snížení ostrosti vidění v 66,7% případů, je přední lenticonus - 37,8%, skvrny na sítnici - v 22,2%, šedý zákal - 20%, keratokonus - 6 , 7%.
U některých dětí s dědičnou nefritidou, zejména při tvorbě renální insuficience, je zaznamenáno významné zpoždění ve fyzickém vývoji. Vzhledem k tomu, že progrese renální insuficience vyvolává hypertenzi. U dětí je častěji zjištěna v dospívání a ve vyšších věkových skupinách.
Vyznačující se tím, že se u pacientů s dědičnou různými nefritida (5-7) stigmat pojivové dizembriogeneza. Mezi pojivové tkáně stigmatu u pacientů s nejčastější oční hypertelorismem, vysoká patra, malocclusion, abnormální tvar uší, zakřivení malíčku na ruce, „sandalevidnaya gap“ na nohou. Pro hereditární nefritida se vyznačuje jednotností dizembriogeneza stigma v rámci rodiny, stejně jako vysoké četnosti jejich distribuce v probandů příbuzných, přes který je přenášena onemocnění.
V časných stádiích onemocnění odhalilo izolovanou snížení funkce ledvin částečného: transport aminokyselin, elektrolytů, koncentrace funkce acidogeneze, další změny jsou funkční stav jak proximální a distální nefronu a mají charakter kombinovaných dílčích poruch. Snížení glomerulární filtrace nastane později, častěji v období dospívání. Jak postupuje dědičná nefritida, rozvíjí se anémie.
Tak, pro hereditární nefritida vyznačující stádia onemocnění: první latentní fázi nebo skryté klinických symptomů projevují minimální změny močového měchýře pak dochází postupný proces dekompenzace se snížením funkce ledvin se zjevnými klinickými symptomy (intoxikace, astenie, opožděný vývoj, anemizatsiya). Klinické symptomy se objevují obvykle bez ohledu na stratifikaci zánětlivé reakce.
Dědičná nefritida se může projevit v různých věkových obdobích, což závisí na působení genu, který je do určité doby v potlačeném stavu.
Klasifikace
Existují tři varianty dědičné nefritidy
- I - klinicky se projevuje nefritidou s hematurií, ztrátou sluchu a poškozením oka. Průběh nefritidy je progresivní při vývoji CRF. Druh dědičnosti je dominantní, spojený s chromozómem X. Morfologicky dochází k narušení struktury bazální membrány, ke ztenčení a štěpení.
- II - klinicky se projevuje nefritidou s hematurií bez ztráty sluchu. Průběh nefritidy je progresivní s vývojem chronického selhání ledvin. Druh dědičnosti je dominantní, spojený s chromozómem X. Morfologicky dochází ke ztenčení bazální membrány glomerulárních kapilár (zejména laminadensy).
- III - benigní rodinná hematurie. Kurz je příznivý, chronické selhání ledvin se nevyvíjí. Druh dědičnosti je autosomálně dominantní nebo autosomálně recesivní. V autozomálně recesivním druhu dědičnosti mají ženy závažnější průběh onemocnění.
Diagnóza Alportova syndromu
Jsou navržena tato kritéria:
- přítomnost v každé rodině alespoň u dvou pacientů s nefropatií;
- hematurie jako hlavní symptom nefropatie ve probandu;
- alespoň jeden člen rodiny má sluchovou ztrátu;
- rozvoj chronického selhání ledvin u jednoho příbuzného a více.
V diagnostice různých dědičných chorob a vrozených důležité místo patří k integrovanému přístupu k inspekci a především pozor na základě údajů získaných při přípravě původu dítěte. Diagnóza syndrom Alportova za platné v případě, že pacient 3 ze 4 Charakteristické vlastnosti: přítomnost v rodině hematurie a chronického selhání ledvin, přítomnost sensorineurální ztráty sluchu pacienta, patologie detekce v elektronových mikroskopických charakterizace biopsie známek štěpení glomerulární bazální membrány se změnou jeho tloušťky a nerovnostem.
Zkoumání pacienta by mělo zahrnovat klinické genetické metody vyšetřování; zaměřené na anamnézu onemocnění; obecné vyšetření pacienta s přihlédnutím k diagnostickým kritériím. Kompenzace etapa patologie mohou chytit pouze se zaměřením na tyto syndromy, že mají rodinnou anamnézu, hypotenze, více blizny dizembriogeneza mění močového měchýře. V dekompenzací estrarenalnyh může způsobit příznaky, jako jsou těžké intoxikace, astenie, retardovaný tělesný rozvoj anemizatsiya projevuje a amplifikace s postupným poklesem funkce ledvin. U většiny pacientů s poklesem funkce ledvin je pozorován pokles funkce acido- a aminogeneze; u 50% pacientů došlo k významnému poklesu sekreční funkce ledvin; omezení rozsahu fluktuací optické hustoty moče; porušení rytmu filtrace a následné snížení glomerulární filtrace. Stupeň chronické selhání ledvin je diagnostikována tím, že se u pacientů po dobu 3-6 měsíců nebo déle, zvýšené hladiny močoviny v séru (více než 0,35 g / l), snižuje glomerulární filtraci až o 25% normálu.
Diferenciální diagnostika dědičné nefritidy musí být provedena především s získaná forma hematuric glomerulonefritidy. Získal stále akutní glomerulonefritidu začínající dobu 2-3 týdnů po předchozím infekci, extrarenální vlastnosti, včetně hypertenze se v prvních dnech (v dědičném nefritidy, naopak, hypotenze), snížená rychlost glomerulární filtrace u počátku, žádné porušení dílčích trubkových funkcí, zatímco jako v tomto dědičné. Získané glomerulonefritidy dochází s těžším hematurie a proteinurie, se zvýšenou ESR. Diagnostické hodnoty jsou typické změny v glomerulární bazální membráně, charakteristická dědičná nefritida.
Diferenciální diagnostika dysmetabolický nefropatie provedena s chronickým selháním ledvin v rodině identifikované klinicky monotyp onemocnění ledvin, a může být v rozmezí od nefropatie pyelonefritidy do urolitiázy. Děti mají často stížnosti na bolest břicha a pravidelně močením v sedimentu moče - oxalátu.
Máte-li podezření, že by pacient měl dědičnou nefritidu, měl by být pacient zaslán, aby diagnostiku objasnil ve specializovaném oddělení nefrologie.
Co je třeba zkoumat?
Jaké testy jsou potřeba?
Kdo kontaktovat?
Léčba Alportova syndromu
V režimu stanoví omezení velké fyzické námahy, pobyt na čerstvém vzduchu. Dieta je vysoce kvalitní s dostatečným obsahem vysoce kvalitních bílkovin, tuků a sacharidů s přihlédnutím k funkci ledvin. Velký význam má identifikace a rehabilitace chronických ložisek infekce. Z léků se používá ATP, kokarboxyláza, pyridoxin (až do 50 mg / den), chlorid karnitinu. Kurzy se konají 2-3x ročně. Když je hematurie předepsána fytoterapie - kopřivovitá, kopřivovitá, černý popel, řebříček.
V zahraniční a domácí literatuře jsou zprávy o léčbě prednisolonem a použití cytostatiky. Efekt je však těžké posoudit.
Při chronickém selhání ledvin se používá hemodialýza a transplantace ledvin.
Neexistují žádné metody specifické (účinné patogenetické) terapie dědičných nefritid. Všechna lékařská opatření směřují k prevenci a zpomalení snížení funkce ledvin.
Dieta by měla být vyvážená a vysoce kalorická, s přihlédnutím k funkčnímu stavu ledvin. Při absenci porušení funkčního stavu ve výživě dítěte by měl být dostatečný obsah bílkovin, tuků a sacharidů. Při přítomnosti příznaků renální dysfunkce by množství bílkovin, uhlohydrátů vápníku a fosforu mělo být omezeno, což zpomaluje vývoj chronického selhání ledvin.
Fyzický stres by měl být omezen, děti by se měly vyhýbat sportu.
Vyhněte se kontaktu s infekčními pacienty, snižte riziko vzniku akutních respiračních infekcí. Je třeba sanovat ohnisky chronické infekce. Preventivní očkování dětí s dědičnou nefritidou se nevykonává, vakcinace je možná pouze podle epidemiologických indikací.
Hormonální a imunosupresivní terapie v dědičné nefritidě je neúčinná. Existují náznaky určitého pozitivního účinku (snížení hladiny proteinurie a zpomalení progrese onemocnění) s dlouhodobým užíváním inhibitorů cyklosporinu A a ACE po mnoho let.
Při léčbě pacientů užívajících léky, které zlepšují metabolismus:
- pyridoxin - 2-3 mg / kg / den ve 3 rozdělených dávkách po dobu 4 týdnů;
- kokarboksilaza - 50 mg intramuskulárně každý druhý den, pouze 10-15 injekcí;
- ATP - 1 ml intramuskulárně každý druhý den, 10-15 injekcí;
- Vitamin A - 1000 U / rok / den v 1 recepci po dobu 2 týdnů;
- vitamín E - 1 mg / kg / den v 1 příjmu po dobu 2 týdnů.
Tato terapie zlepšuje celkový stav pacientů, snižuje tubulární dysfunkci a podává se 3krát ročně.
Jako imunomodulátor lze užívat levamizol - 2 mg / kg / den 2-3x týdně s přerušeními mezi dávkami 3-4 dnů.
Výzkumníkům má hyperbarická okysličení pozitivní vliv na závažnost hematurie a renální dysfunkce.
Nejúčinnějším způsobem léčby dědičných nefritid je včasná transplantace ledvin. Pokud to tak není pozorován v transplantační relapsu, u malého procenta (asi 5%) může nephritis vývoj v transplantované ledviny, spojené s antigeny v glomerulární bazální membráně.
Slibnou oblastí je prenatální diagnostika a terapie genetického inženýrství. Pokusy na zvířatech ukazují vysokou účinnost přenosu normálních genů odpovědných za syntézu a-řetězců kolagenu typu IV do ledvinového tkáně, po němž se pozoruje syntéza normálních struktur kolagenu.
Předpověď počasí
Prognóza dědičné nefritidy je vždy vážná.
Prognosticky nepříznivé kritéria pro tok dědičných nefritid jsou:
- mužský sex;
- včasný rozvoj chronického selhání ledvin u členů rodiny;
- proteinurie (více než 1 g / den);
- zahušťování glomerulární bazální membrány podle mikroskopie;
- neuritis sluchového nervu;
- delece v genu Co4A5.
Prognóza benigní rodinné hematurie je výhodnější.
Использованная литература