
Alkaptonurie - vrozená enzymatická patologie
Naposledy posuzováno: 23.04.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
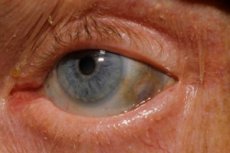
Jedna z velmi vzácných metabolických poruch - alkaptonurie - se týká vrozených abnormalit v metabolismu aminokyseliny tyrosinu.
Tento syndrom lze také nazvat deficit homogentisát oxidázy, homogentisinurie, dědičná ochronóza nebo onemocnění černé moči. [1]
Epidemiologie
Podle statistik není více než devět případů alkaptonurie na 1 milion populace. A ve většině evropských zemí - jeden případ na 100–250 tisíc živě narozených.
Mezi evropskými zeměmi je výjimkou Slovensko (zejména relativně malý severozápadní region), kde je prevalence alkaptonurie jedním případem u 19 tisíc novorozenců. S největší pravděpodobností je to dáno skutečností, že mezi rodinami slovenských Romů, kteří tam žijí, je míra příbuzenského křížení (manželství mezi bratranci a sestřenicemi) nejvyšší v Evropě: 10–14%. [2]
Příčiny alkaptonurie
Přesné příčiny alkaptonurie, jako vrozené poruchy katabolismu (metabolické poruchy) aromatického (homocyklického) α-aminokyselinového tyrosinu, byly stanoveny: tento typ metabolické poruchy je důsledkem homozygotních nebo komplexních heterozygotních mutací jedné z tisíců genů na chromozomu 3, přesněji gen HqG21 v lokusu 3 -q23 na dlouhém rameni chromozomu. Tento gen kóduje nukleotidové sekvence jaterního enzymu homogentisate-1,2-dioxygenázy [3](také nazývaného oxidáza homogentisové kyseliny nebo homogentisát oxidázy), metaloproteinu obsahujícího železo potřebného pro jeden ze stupňů rozpadu tyrosinu v těle. [4], [5]
Alkaptonurie je tedy defektem enzymu homogentisát-1,2-dioxygenázy, přesněji důsledkem jejího geneticky podmíněného nedostatku nebo úplné absence. [6]
Alkaptonurie je vrozená fermentopatie dědičná jako autozomálně recesivní znak, to znamená, že aby se alkaptonurie mohla vyskytnout u dětí, musí mít oba rodiče upravený gen pro tento enzym, protože každý z nich přenáší na dítě pouze jednu kopii gen ze dvou dostupných.
Podle nejnovějších údajů existuje více než dvě stě variant modifikace genu HGD a nejčastěji jsou pozorovány mutace missense, translokace a sestřih.
Rizikové faktory
Jediným rizikovým faktorem vzniku této vrozené fermentopatie je její rodinná anamnéza a dědičnost dvou modifikovaných kopií genu HGD, pokud rodiče nevykazují alkaptonurii (riziko přenosu anomálie je 25%), nebo jednu z rodiče mají tuto poruchu. [7]
Patogeneze
Nejdůležitější roli hraje tyrosin při syntéze bílkovin, produkci chromoproteinů - kožního pigmentu melaninu, dále hormonů štítné žlázy a katecholaminových neurotransmiterů.
Mechanismus regulace množství tyrosinu v buňkách je velmi složitý a tělo svůj přebytečný obsah normalizuje štěpením. Proces katabolismu tyrosinu, stejně jako všechny aromatické aminokyseliny, je vícestupňový a probíhá v několika fázích. Navíc každý stupeň metabolického rozkladu tyrosinu probíhá za účasti určitého enzymu a tvorby meziproduktu.
Aminokyselina se tedy nejprve štěpí na para-hydroxyfenylpyruvát, který se změní na alkapon-2,5-dihydroxyfenyloctovou nebo homogentisovou kyselinu. Dále by měl být alkapon přeměněn na kyselinu maleoctovou, ale to se nestává. [8]
A patogeneze alkaptonurie spočívá v zastavení biochemických reakcí tyrosinového katabolismu ve stádiu tvorby homogentisové kyseliny: k jejímu rozkladu jednoduše neexistuje žádný potřebný enzym - homogentisát oxidáza.
Tělo kyselinu homogentisovou nepoužívá a může se hromadit vylučováním ledvinami. Kromě toho se oxiduje na benzochinoacetát (kyselina benzochinonoctová), který vazbou na molekuly tkání a tělních tekutin vytváří biopolymerní sloučeniny zbarvené jako melanin.
Akumulace těchto meziproduktů ve tkáni vede k narušení struktury kolagenu chrupavkové tkáně, v důsledku čehož klesá její pružnost - s výskytem mnoha klinických příznaků alkaptonurie a rozvojem komplikací.
Symptomy alkaptonurie
Alcaptonurie u novorozenců a kojenců se projevuje ztmavnutím moči. Při kontaktu se vzduchem se barva moči na plenkách, plenkách a spodním prádle změní na tmavě hnědou; je to způsobeno akumulací a uvolňováním kyseliny homogentisové, která se oxiduje na benzochinoacetát. [9]
Při absenci dalších příznaků není alkaptonurie u dětí v raném věku často rozpoznána včas, protože po močení může moč po několika hodinách ztmavnout. Podle některých zpráv je v podmínkách klinik detekována pouze jedna pětina dětí mladších 12 měsíců, které se narodily s touto fermentopatií. Proto je pro rodiče velmi důležité věnovat péči o dítě.
Počáteční příznaky navíc zahrnují pigmentaci (modravě šedou) oční skléru a chrupavku ušních boltců a nosu, často označovanou jako ochronóza. [10]
Časem se objevují další příznaky:
- silná pigmentace kůže na lícních kostech, podpaží a genitáliích;
- barvení oděvů při kontaktu s pocujícími částmi těla;
- záchvaty obecné slabosti;
- chraplák.
Je třeba mít na paměti, že alkaptonurie a ochronóza , jak bylo uvedeno výše, jsou synonymní názvy pro stejné porušení tyrosinového katabolismu.
Nemoc z javorového sirupu a alkaptonurie. Vrozená choroba javorového sirupu nebo leucinóza také označuje metabolické poruchy, má stejný typ dědičnosti a dokonce se vyskytují mutace na stejném chromozomu, ale týkají se genu kódujícího komplex enzymů rozvětvené a-keto kyseliny dehydrogenázy. Z tohoto důvodu nemůže tělo rozbít určité proteinové složky, zejména aminokyseliny leucin, izoleucin a valin. V tomto stavu má moč (a ušní maz) sladkou vůni; v klinickém obrazu tohoto typu organické acidémie je navíc pozorována hypopigmentace, kolísání krevního tlaku, křeče, zvracení a průjem, pokles hladiny glukózy v krvi, ketoacidóza, halucinace atd. Úmrtnost u dětí je poměrně vysoká vysoký; u dospělých bez léčby může v důsledku mozkového edému dojít ke kómatu a smrti.
Albinismus a alkaptonurie „spojují“ pouze tyrosin. Albinismus , včetně okulokutánního albinismu, je způsoben genetickými mutacemi, které ovlivňují produkci pigmentového melaninu. Vrozené změny jsou zaznamenány v genu TYR na chromozomu 11 (11q14.3), který kóduje tyrosinázu, enzym obsahující měď v melanosomech, který je nezbytný pro tvorbu kožního pigmentu na základě produktů metabolismu tyrosinu. Toto onemocnění je mnohem častější než alkaptonurie.
Komplikace a důsledky
Následky a komplikace alkaptonurie způsobené působením přechodných metabolitů tyrosinu - homogentisové a benzochinonoctové kyseliny - se objevují v důsledku ukládání reaktivních pigmentovaných polymerů, destrukci kolagenových fibril a zhoršování stavu chrupavky (s poklesem jejich odolnosti) na mechanické namáhání).
O několik let později, v dospělosti, se vyvíjí degenerativní artritida a osteoartróza velkých kloubů (kyčel, sakroiliak a koleno); zúžení meziobratlových prostorů (zejména bederní a hrudní páteře) - s kalcifikací a tvorbou osteofytů; hustota tkáně subchondrálních kostních plotének klesá a podkladové kosti mohou projít patologickou remodelací s tvorbou výrůstků a deformací. [11]
Může dojít k poškození srdečních chlopní (aortální a mitrální) a koronárních tepen se známkami ischemické choroby srdeční a také tvorbě ledvinových a prostatických kamenů v důsledku stejné kalcifikace. [12], [13]
Diagnostika alkaptonurie
Diagnóza vrozených metabolických poruch je obvykle založena na studiu tělesných tekutin.
Na základě jakých testů a jakých reakcí lze diagnostikovat alkaptonurii? Jsou vyžadovány testy moči - pro detekci kyseliny homogentisové a stanovení její hladiny (normální - 20-30 mg denně, zvýšené - 3-8 g). Vzorek moči se zkouší plynovou chromatografií nebo hmotnostní spektrometrií pomocí kapalinové chromatografie; je možný screeningový test na přítomnost chloridu železitého v moči. [14]
Existuje také rychlá diagnostická metoda - stanovení alkaptonu v zaschlých skvrnách moči na papíře (podle intenzity barvy).
Při objasňování diagnózy instrumentální diagnostika (rentgen) zahrnuje identifikaci rentgenových příznaků osteoartrózy a dalších kloubních patologií u pacientů.
Diagnóza je potvrzena takovými& molekulárně genetickými metodami, jako je diagnostika dědičných chorob , jako je genetické testování a sekvenování DNA. [15]
Diferenciální diagnostika
Diferenciální diagnostika zahrnuje hemochromatózu a akutní selhání jater novorozenců, melaninurii, akutní intermitentní porfyrii, hemofagocytární lymfohistiocytózu, primární mitochondriální patologii, revmatoidní artritidu, ankylozující spondylitidu.
Kdo kontaktovat?
Léčba alkaptonurie
Hlavní léčbou alkaptonurie je požití velkých dávek (nejméně 1 000 mg denně) kyseliny askorbové. U dětí to zvyšuje vylučování kyseliny homogentisové močí a u dospělých snižuje hladinu jejího derivátu, kyseliny benzochinonoctové, v moči a zpomaluje její vazbu na struktury pojivové tkáně kloubů a kolagenu. [16]
Na západoevropských klinikách se testuje lék Nitizinone (Orfalin), lék ze skupiny metabolitů, který inhibuje druhý stupeň tyrosinového katabolismu: transformaci para-hydroxyfenylpyruvátu na kyselinu homogentisovou. Použití tohoto farmakologického činidla však vede k akumulaci tyrosinu a může způsobit závažné vedlejší účinky, včetně neprůhlednosti rohovky a fotofobie, krvácení z nosu a žaludku, selhání jater, změny v krvi atd. Ve Spojených státech však Nithizinone je schválen FDA pro léčbu tyrosinémie typu I. [17], [18]
Pro problémy s klouby způsobené alkaptonurií se jako taková provádí fyzioterapie - cvičební terapie ke zvýšení svalové síly a zvýšení pohyblivosti kloubů, balneo a peloidoterapie k omezení bolesti.
Přestože tyrosin není dodáván pouze s jídlem, ale je také produkován v těle, pacientům s alkaptonurií se doporučuje dieta s nízkým obsahem bílkovin a omezující příjem potravin bohatých na tyrosin, především hovězí a vepřové maso, mléčné výrobky (zejména sýry), luštěniny, ořechy atd. Semena.
Prevence
Prevence genových mutací je nemožná a aby se zabránilo narození dětí s vysokým rizikem vrozených poruch, existuje lékařské a genetické poradenství, které je nutné před plánovaným těhotenstvím těch manželských párů, v jejichž rodinné anamnéze se vyskytují dědičné choroby Příroda. [19]
Předpověď
V průběhu alkaptonurie existuje jen velmi málo úmrtí a smrt může být způsobena vážnými komplikacemi postihujícími srdce a ledviny. Pro celkovou délku života lidí s alkaptonurií je tedy prognóza příznivá.
Ale kvalita života je snížena kvůli intenzivní bolesti kloubů nebo páteře s výrazným omezením pohyblivosti, často progresivní.