
Angelmanův syndrom u dětí a dospělých
Naposledy posuzováno: 23.04.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.

Existuje řada onemocnění, při nichž výrazy jako "starat se o sebe a nemusel se zvracet", alespoň směšný. Tato patologie, při níž jsou v těle dítěte uloženy některé mentální a fyzické abnormality ještě před narozením, ale rodiče nemají žádnou vinu. Taková onemocnění jsou způsobena mutacemi nebo poruchami v sadách chromozomů a jsou nazývána chromozomální nebo genetickou. Angelmanův syndrom, Downův syndrom, Patau, Edwards, Turner, Prader-Willi jsou jen součástí genetických onemocnění z poměrně slušného seznamu.
Syndrom šťastné osoby
Tentokrát budeme mluvit o nemoci, pojmenoval podle britského pediatr Harry Angelmanovým od první otázku vyvolal na problém z roku 1965 rok, stál v předvečer své praxi se třemi neobvyklými dětmi, sjednocená společnými zvláštních příznaků. Lékař jmenoval tyto dětské loutkové děti a napsal o nich článek, který byl původně nazván "Loutkové děti". Samotný článek a jeho jméno byly napsány pod dojmem obrazu v jednom z muzeí ve Veroně. Na obrázku se zobrazoval chlapík se smíchem, a to bylo nazváno "Boy-loutka". Sdružení dítěte zobrazené na obrázku se třemi dětmi, které Angelman kdysi narazil ve své praxi, a povzbuzoval pediatra, aby sjednotil děti v jedné skupině kvůli jejich stávající nemoci.
Skutečnost, že děti uvedené v článku nebyly zaznamenány jinými lékaři, není překvapující. Koneckonců na první pohled se zdálo, že mají zcela odlišné nemoci, takže obecný klinický obraz onemocnění se lišil ve třech různých případech. Nová "chromozomální patologie může být zajímavá i pro další vědce, ale v té době nebyla genetika dostatečně rozvinutá, aby potvrdila hypotézu anglického lékaře. Proto se článek po jistém zájmu o něj dlouho opustil vzdáleného pluku.
Další zmínka o Angelmanově syndromu, a tak se nyní nazývala článek pediatra z Anglie, G. Anglemanna, pochází z počátku 80. Let 20. Století. A teprve v roce 1987 bylo možné najít důvod, proč se taková odchylka rodí malá část dětí, že ze strany vypadají neustále usměvavá a šťastní. Ve skutečnosti to není tak, a úsměv je jen grimasa, za nímž spočívá nešťastná lidská duše a bolest rodičů.
Epidemiologie
Chromozomální mutace u dítěte se může podle statistik vyvíjet jak na pozadí takových mutací u rodičů, tak v případě, že taková mutace neexistuje. V Angelmanově syndromu (SA) není jasný dědičný charakter, ale pravděpodobnost vzniku patologie u rodičů s chromozomálními mutacemi je poměrně vysoká.
Je také zajímavé, že pokud rodina již má dítě s SA, existuje jeden procentní příležitost mít druhé dítě stejného druhu, i když jsou rodiče zdraví.
Dosud neexistují přesné statistické údaje o počtu pacientů s Anghelmanovým syndromem. Možná chyba je řada příznaků, které se mohou vyskytnout v určitém složení nebo dlouho nevyplývají vůbec. Předpokládá se, že prevalence onemocnění je: 1 dítě na 20 000 novorozenců. Ale toto číslo je velmi přibližné.
Příčiny angelmanův syndrom
Angelmanův syndrom je lékařský název pro chromozomální patologii, ale není to ani jediný. U lidí, nemoc se nazývá i loutkové syndrom děti, syndrom happy loutkové a syndrom petržel a smáli syndrom panenky. Ano, některé názvy prostě nemají přijít s lidmi (někdy dokonce urážky na samotných i jejich rodiče pacientů), ale nemoc je nemoc, jaké by to bylo zábavné, nebo se podíval ven a to bez ohledu na důvody mohou být způsobeny.
Důvody vzniku Angelmanova syndromu a mnoho dalších genetických patologií jsou ve všech případech porušení struktury jednoho z chromozomů nebo celé chromozomové soustavy. Jenom v našem případě však celý problém spočívá v 15 chromozomech přenášených od matky. Tedy. Rodičovský chromozóm nemá v tomto případě žádné odchylky, ale žena prochází určitými mutacemi.
Podle typu chromozomální abnormality se Angelmannův syndrom týká chromozomálních mutací. Takové mutace jsou:
- Vypuštění (nedostatek oblasti chromozomu, který obsahuje specifickou sadu genů, a není-li jeden z genů jde mikrodelece), což je výsledkem dvou nespojitosti a jednoho sloučení když ztratil část původního chromozomu.
- Duplikace (přítomnost dalšího místa v chromozomu, což je kopie již dostupného), což ve většině případů vede k smrti člověka, méně často k neplodnosti.
- Inverzní (inverze chromozomu částí o 180 °, to znamená, v opačném směru, a pak geny v nich jsou uspořádány v opačném pořadí), kdy končí přerušovaná chromozóm spojené v jiném pořadí, než původní.
- Vložení (pokud část genetického materiálu v chromozomu není na jeho místě),
- translokace (pokud část chromozomu spojí další chromozom, taková mutace může být vzájemná bez ztráty míst).
Získat mutovaný chromozóm od netušící matky, dítě je odsouzeno k tomu, že se narodí s odchylkami předem. Nejčastější příčinou vzniku Anghelmanova syndromu je stále vymazání mateřského chromozomu 15, kdy v něm není malá oblast. Méně často se vyskytující mutace v syndromu "smíchu panenky" jsou:
- translokace,
- jednoosá diosomie (jestliže dítě dostalo od otce pár chromozomů, mateřský chromozom chybí)
- mutace genů v DNA, které jsou jak hlavním materiálem stavebního materiálu (genetický), tak i instrukcí pro jeho správné použití (zejména mutace genu ube3a v mateřském chromozomu).
Přítomnost jedné z těchto mutací u rodičů je rizikovým faktorem Anghelmanova syndromu u dětí. Ale nejen chromozomální mutace, ale genom (které jsou spojeny s kvantitativní změny v chromozomu sad a běžnější chromozomu) může vyvolat vývoj onemocnění u dítěte. Ke společným genomovým mutacím lze přičíst trisomii chromozomů (pokud má osoba chromozomální soubor s více než 46 chromozomy).
K patologii dítěte nemusí nutně mít rodiče chromozomální abnormality. A přesto existuje určité procento pacientů, jejichž onemocnění je dědičné.

Patogeneze
Něco ponořme do biologie, přesněji do genetiky. Genetická informace každého jednotlivého lidského těla je obsažena ve 23 párech chromozomů. Jeden chromozom z páru je přenášen na dítě od otce, druhý od matky. Všechny páry chromozomů se liší ve tvaru a velikosti a obsahují určité informace. Takže 23 párů chromozomů (chromozómy X a Y) je zodpovědný za formování sexuálních charakteristik dítěte (XX - dívka, XY-chlapec, zatímco dítě může dostat Y-chromozom pouze od svého otce).
V ideálním případě dítě dostává od svých rodičů 46 chromozomů, které tvoří jeho genetické atributy a předurčují ho jako jednotlivce. Vyšší počet chromozomů se nazývá trisomie a považuje se za odchylku od normy. Například přítomnost 47 chromozomů v chromozomovém souboru (karyotyp, který určuje druh a individuální charakteristiky) způsobuje nástup Downovho syndromu.
Pokud jsou chromozomy zbarveny zvláštním barvivem, pak v mikroskopu vidíme kapely různých odstínů podél každého z nich. V každé skupině je obrovský počet genů. Všechny tyto kapely jsou očíslovány vědci a mají pevné místo. Absence jedné z pásem je považována za odchylku od normy. Při Angelmannově syndromu můžete velmi často pozorovat nepřítomnost segmentů mateřského chromozomu v intervalu q11-q13 umístěném v dlouhé paži, počet DNA základen, ve kterých je jen asi 4 miliony.
Hlavní složkou chromozomu je neuvěřitelně dlouhá molekula DNA obsahující tisíce genů a desítek a stovek milionů dusíkatých bází. Takže 15 chromozomů zodpovědných za vývoj Angelmanova syndromu a několik dalších obsahuje 1200 genů a asi 100 milionů základů. Jakékoli porušení struktury molekuly DNA nutně ovlivní vzhled a vývoj nenarozeného dítěte.
Genetická informace obsažená v genech se převádí na protein nebo RNA. Tento proces se nazývá genová exprese. Genetická informace získaná od rodičů tedy získá formu i obsah, ztělesněný v jejich jedinečném dědicu ženského nebo mužského pohlaví.
Existuje řada chorobných stavů s neklasických typu dědičnosti, včetně Angelmanovým syndrom, u kterého jsou geny obdržel od rodičů jako součást spárovaných chromozomů unikátní otisk rodiče a projevují různými způsoby.
Takže, Angelmanův syndrom je ukázkovým příkladem genomového imprintingu, přičemž exprese genů v těle dítěte je přímo v závislosti na z nichž mateřských odvozený alely (různé formy téhož genu získaného z otce a matky jsou umístěny na stejných částech párových chromozomů) . Tedy. Vede ke vzniku syndromu abnormalit v mateřské chromozomu, zatímco mutace a poruchy otcovští struktura chromosomu způsobit velmi různých chorob.
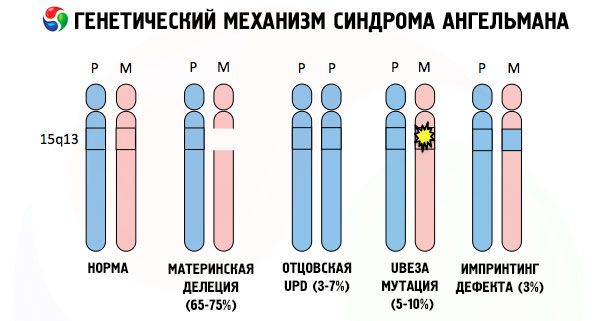
Při této nemoci je nedostatek specifických genů v mateřském chromozomu nebo ztráta / snížení aktivity jednotlivých genů (ve většině případů ube3a gen podílející se na metabolismu ubikvitin - degradace proteinů jiné regulační proteiny). Výsledkem je, že dítě je diagnostikováno s abnormalitami duševního vývoje a fyzickými deformacemi.
Symptomy angelmanův syndrom
Symptomatologie Angelmanova syndromu ovlivňuje různé aspekty života a vývoje dítěte: fyzické, neurologické a psychické. Na základě toho můžeme rozlišovat tři skupiny příznaků, které naznačují vývoj této patologie.
- Externí nebo fyzické příznaky:
- neúměrně malou hlavu ve srovnání s kmenem a končetinami, které mají normální velikost,
- příliš široká ústa,
- na tváři je téměř vždy úsměv (s otevřenými ústy),
- vzácné zuby,
- úzký horní ret,
- často vytáhne široký jazyk,
- vyčnívající spodní čelist,
- ostrá brada,
- velmi lehká kůže, často také vlasy (albinismus, spojený se skutečností, že tělo produkuje pigmentový melanin),
- tmavé skvrny na lehké pokožce (hypopigmentace v důsledku nedostatečné produkce melaninu)
- fyzické nebo vnější příznaky: oční onemocnění, jako je strabismus nebo atrofie optického nervu,
- zakřivení páteře (skolióza),
- ztuhlé nohy (při chůzi člověk neohýbá kolena kvůli malé pohyblivosti kloubů, tudíž i srovnání s loutkovou chůzí).
- Symptomy spojené s mentálním a emočním vývojem:
- silné zpoždění v duševním vývoji,
- zbytečně emocionální, hlučné, rozrušené chování,
- časté tleskání,
- vyjádřená vstřícnost, zdůrazněná konstantním úsměvem na obličeji,
- častý, bezvýznamný smích.
- Neurologické příznaky:
- třesení končetin,
- nedostatečná koordinace pohybů se ztrátou rovnováhy,
- snížený svalový tonus,
- různé poruchy spánku,
- časté hysterické záchvaty v dětství,
- porucha řeči (dítě začíná mluvit pozdě, má špatné komunikační dovednosti a mrzutý projev),
- hyperaktivita na pozadí zvýšené excitability,
- problémy se soustředěním a tréninkem.
Ale toto je zobecněný obraz této nemoci. Ve skutečnosti klinický obraz Anghelmanova syndromu do značné míry závisí na stupni vývoje onemocnění a typu chromozomální mutace, která způsobila patologii. A to znamená, že u různých pacientů se symptomologie onemocnění může významně lišit, což již po dlouhou dobu neumožnilo izolovat patologii mimo jiné s podobným klinickým obrazem.
Z celkového počtu symptomů lze identifikovat takové, které jsou charakteristické pro všechny pacienty bez výjimky:
- závažné odchylky v duševním vývoji,
- nedostatečné chování (bezdůvodný smích, zvýšená excitabilita, špatná koncentrace pozornosti, stav euforie),
- nedostatečné rozvoj motorických dovedností,
- špatná koordinace pohybů, ataxie chůze (nerovnoměrné tempo, kývání ze strany na stranu atd.), třesení končetin.
- porušení vývoje řeči s převahou nonverbálních komunikačních prostředků.
Mezi příznaky, které se vyskytují u drtivé většiny pacientů, můžeme rozlišit:
- nepřiměřená hlava a kmen, způsobené zpožděním ve fyzickém vývoji,
- u mnoha pacientů je tvar lebky takový, že velikost mozku zůstává menší než u zdravých lidí (mikrocefalie),
- epileptické záchvaty ve věku do 3 let s progresivním poklesem síly a frekvence u starších pacientů,
- zkreslení indexů EEG (oscilace a vysoká amplituda nízkofrekvenčních vln).
Tyto příznaky se vyskytují poměrně často, přesto u 20% pacientů s Angelmannovým syndromem chybí.
Ještě méně zřídka může diagnostikovat takové projevy nemoci jako:
- výrazný nebo mírný strabismus,
- slabá kontrola nad pohybem jazyka, v důsledku čehož pacienti často vyčnívají z jazyka bez důvodu,
- potíže s polykáním a sání, zejména u mladších dětí,
- porušení pigmentace kůže a očí,
- zvednutý nebo ohnutý v průběhu chůze,
- giperperflexie,
- poruchy spánku, zejména v dětství,
- časté salivace,
- nepotlačitelná žízeň,
- nadměrně aktivní žvýkací pohyby,
- přecitlivělost na teplo,
- plochá hlava,
- pokročilá dolní čelist,
- hladké dlaně.
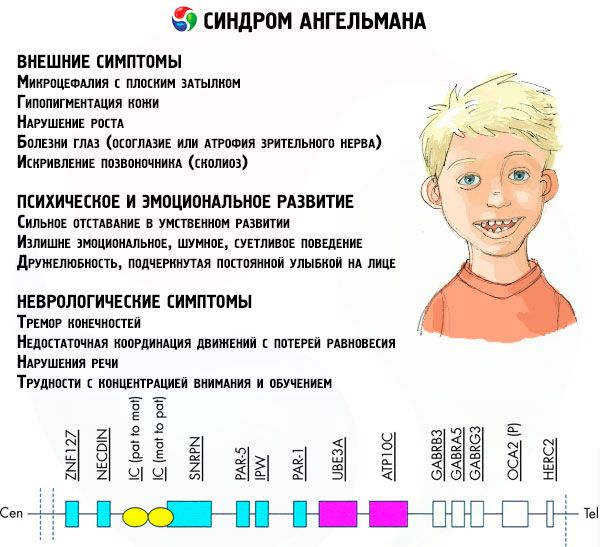
Poměrně velké procento pacientů má problémy s močením, které špatně kontrolují, porušení jemných motorických dovedností, což vytváří potíže při samoobsluze a výcviku, nadváhou. Prakticky u všech pacientů začne puberta později než u zdravých vrstevníků.
Děti s angelmanovým syndromem jsou dobré při porozumění a porozumění ústnímu projevu, ale nechtějí se účastnit rozhovoru, omezujícím projev na desítky slov nezbytných v každodenním životě. V dospělosti však tito pacienti vypadají mladší než jejich rodiče bez genetických patologií.
Mnohé z příznaků Angelmannova syndromu jsou nestálé, takže klinický obraz nemoci se mění s věkem. Záchvaty a epileptické záchvaty se stávají vzácnějšími nebo vůbec zmizí, pacient se stává méně nervózní,
Komplikace a důsledky
Angelmanův syndrom je vážná, prakticky nevyléčitelná až dosud chromozomální patologie, která pacientům zbavuje příležitosti žít v normálním životě. Jaký bude život dítěte s SA, do značné míry závisí na typu chromozomální abnormality.
Duplikace oblasti chromozomu je ve většině případů nekompatibilní se životem. A i kdyby tito pacienti nezemřeli v dětství a nedosáhli puberty, nemají možnost mít děti.
Vymazání nebo nepřítomnost části genů, ke kterým dochází u Anghelmanova syndromu, je nejčastěji překážkou pro dítě, které se učí chodit a mluvit. U takových dětí dochází mentální retardace v závažnější formě, objevují se často epileptické záchvaty a jejich intenzita je mnohem silnější než u pacientů s jinými chromozomálními abnormalitami.
Jestliže je mutace jednoho genu, s přihlédnutím a přístupu, dítě může naučit základy samoobslužné komunikace a komunikace v týmu, i když to bude ještě zaostávají ve vývoji svých vrstevníků.
Pro děti s Angelmannovým syndromem, dobrosrdečný přírodou, hlavní je láska a pozornost rodičů. Pouze v tomto případě výcvik dítěte přinese ovoce, dokonce i malé. Samozřejmě, že pacienti v normální škole nebudou moci studovat s SA. Potřebují speciální třídy, kde se děti nejprve naučí soustředit svou pozornost a potom postupně dávají základy znalostí školy.
Diagnostika angelmanův syndrom
Angelmanův syndrom je vrozená patologie vývoje. Ovšem kvůli jistým okolnostem je diagnostikování v dětství a v raném dětství nejčastěji možné. Důvodem je nespecificita a mírné symptomy u kojenců a batolat až do 3 let. A prevalence onemocnění v naší zemi není tak velká, že se lékaři naučili rozpoznat ji mezi jejími podobnými.
Angelmanův syndrom u dětí se může projevit ve formě snížení svalového tonu, která se projevuje v podobě problémů krmení (slabost sání a polykací reflex) a později se učí jen obtížně chůzi (tyto děti mnohem později začít chodit). Tyto symptomy jsou prvními příznaky odchylky ve vývoji dítěte, což může být spojeno s chromozomální abnormalitou. Potvrďte tento předpoklad pouze genetické analýzy.
Zvláštní pozornost je věnována dětem, jejichž rodiče mají odlišné genomové nebo chromozomální abnormality. Koneckonců, nemoc se nemůže projevit žádným způsobem, a pokud je patologie odhalena včas, tím, že začne tvrdě pracovat s dítětem, je možné dosáhnout mnohem většího úspěchu v tréninku, což zpomaluje průběh onemocnění.
Pokud mají rodiče odlišné chromozomální abnormality, genetická analýza se provádí ještě před narozením dítěte, protože CA je jednou z patologií, které lze zjistit v embryonálním stavu.
Sběr materiálu pro genetický výzkum lze provést dvěma způsoby:
- invazivní (s určitým procentním rizikem, protože je nutné vstoupit do dělohy, aby se podstoupil test plodové vody),
- neinvazivní (DNA analýza dítěte mateřskou krví).
Poté se uskuteční následující výzkumy:
- fluorescenční in situ hybridizaci (metoda FISH) - vazba DNA sondy značené speciálním barvivem na studovanou DNA, následovanou mikroskopickým vyšetřením.
- analýza mutací v genu ube3a a imprinting genů,
- analýza metylace DNA pomocí specifických metod používaných v genetice.
Genetické analýzy poskytují docela přesné informace v případě chromozomálních abnormalit, takže budoucí rodiče předem vědí, na čem by měli být připraveni. Nicméně existují výjimky. U určité skupiny pacientů, za přítomnosti všech příznaků naznačujících příznaky, zůstávají výsledky analýz normální. Tedy. K odhalení patologie je možné jen pečlivě pozorovat dítě od nejranějšího dětství: jak se jí, když začal chodit a mluvit, zda se při chůzi ohýbají nohy, atd.
Kromě FISH-metoda, u metod pro diagnostický nástroj Angelmanův syndrom může být rozlišující tomografie (CT nebo MRI), pomáhá k určení stavu a velikost mozku, a elektroencefalogram (EEG), které ukazují, jak jednotlivé části mozku pracovat.
Konečná diagnóza lékařů je obvykle stanovena ve věku 3-7 let, kdy pacient má již většinu příznaků a je viditelná dynamika vývoje onemocnění.
Jaké testy jsou potřeba?
Diferenciální diagnostika
Angelmanův syndrom je genetická patologie, která ve skutečnosti nemá specifické projevy. Většina symptomů může znamenat jak CA tak další genetické patologie.
Diferenciální diagnóza u Anghelmanova syndromu se provádí s následujícími patologiemi:
- Pitt-Hopkinsův syndrom (pacienti se vyznačují mentální retardací, veselou povahou, usmívají se, mají poměrně velké a široké ústa, je zaznamenána mikrocefalia). Rozdíl - útoky hyperventilace a zpoždění dýchání ve stavu bdění.
- Christianson syndrom (pacienti jsou mentálně zaostalí lidé s veselou povahu, kteří nemohou mluvit, jsou charakterizovány mikrocefalie, ataxie, křeče, mimovolné pohyby svalů).
- Syndrom Mowata-Wilsona (příznaky: mentální retardace, epileptické záchvaty, ostrá brada, otevřené ústa, výraz štěstí na obličeji, mikrocefalie). Rozdíl je velkou vzdáleností mezi očima, oči jsou zkosené dovnitř, špička nosu je zaoblená, ušní část je otočena dozadu.
- Kabuki syndrom (charakteristické mírné až středně těžké mentální retardace, problémy s řečí a motoriky, svalová slabost, záchvaty, mikrocefalie, značné rozdíly mezi zudami, nekoordinovanost). Rozdíl - obočí ve tvaru oblouku, obrácená boční část spodního víčka, široké oči, dlouhé oční štěrbiny s dlouhými hustými řasami.
- Rett syndrom (diferenciace s CA u žen). Symptomy: prodloužený vývoj řeči, křečové záchvaty, mikrocefalie. Rozdíl - na tváři není šťastný výraz, dochází k záchvatům apnoe a apraxie, které nakonec postupují.
- Syndrom autozomálně recesivní duševní tardatsii 38 (příznaky: mentální retardace s nápadným zpožděním ve vývoji motoriky a řeči, svalové slabosti, krmení problémy v dětství, impulzivity). Rozdíl je v modré barvě duhovky.
- Syndrom duplikace genu MESR 2 (diferenciace s SA u mužů). Symptomy: těžká mentální retardace, svalová slabost z dětství, problémy s řečí nebo nedostatek, epilepsie. Rozdíly - progresivní myopatie, neustále se opakující infekce.
- Klifstra syndrom (příznaky: problémy s řečí a myšlení, svalová slabost, poruchy spánku, nedostatku pozornosti, otevřenými ústy, hyperaktivita, záchvaty, ataxie, poruchy rovnováhy). Rozdíly - plochý obličej, krátký zápachový nos, široké oči, velký obrácený spodní ret, útoky agrese.
- Syndrom Smith-Magenis (charakterizovaný záchvaty, problémy se spánkem, poruchy duševního a motorického vývoje). Rozdíly - široká a plochá tvář, konvexní čelo.
- Kulena-de Vriesův syndrom (mírná a středně těžká mentální retardace, svalová slabost, konvulzivní záchvaty, vstřícnost). Rozdíly - dlouhá tvář s vysokým čelem, vyčnívající uši, šikmé oči, větší pohyblivost kloubů, vrozené srdeční patologie.
- Syndrom Philan - McDermid (příznaky: mentální retardace, poruchy řeči nebo jejich nedostatek). Rozdíly - velké ruce s rozvinutými svaly, svalová slabost od narození, slabé pocení.
Angelmanův syndrom podobné symptomy mohou „se mohou pochlubit“ a jak takový nedostatek patologie adenilsuktsinazy, autozomálně recesivní syndrom mentální retardace 1, duplikace syndrom chromozóm 2q23.1, haploinsufficiency geny FOXG1, STXBP1 nebo MEF2C a další.
Úkolem lékaře je provést přesnou diagnózu, diferencovat Angelmannův syndrom od patologických stavů s podobnými příznaky a předepisovat účinnou léčbu, která je relevantní pro diagnostikovaný stupeň vývoje onemocnění.
Léčba angelmanův syndrom
Angelmanův syndrom se odkazuje na kategorii těchto onemocnění, hledání účinného léčení, které léky se zabývají dodnes. Etiologická léčba onemocnění je ve fázi vývoje různých metod a prostředků, z nichž mnohé ještě nebyly testovány u lidí. Doposud lékaři musí být omezena na symptomatickou léčbu pomoci nějak zmírnit utrpení dětí a dospělých s loutkového syndromem, kteří trpí epileptickými záchvaty, slinění, hypotenze a poruch spánku.
Takže snížení frekvence a síly epileptických záchvatů může být u správně vybraného antikonvulzivního léku. Celá obtíž je však, že záchvaty u pacientů s AS se liší od obvyklých epileptických záchvatů tím, že se vyznačují několika typy křečí, což znamená, že bude možné zmírnit stav podáním několika léků najednou.
Nejoblíbenější antikonvulsivní používá k léčbě Angelmanův syndromu jsou: kyselina valproová, topiramát, lamotrigin, levetiracetam, klonazepam a přípravky na bázi těchto. Méně často používají léky na bázi karmazepina, fenytoinu, fenobarbitalu, ethosuximid, protože některé z nich by mohly vyvolat paradoxní efekt, je posílit a zvýšit četnost epileptických záchvatů. K tomu dochází, pokud je léčivo používáno jako součást monoterapie.
Pro léčení slinění se obvykle používají dvě metody: léčivé (přípravky potlačující tvorbu slin) a operační, spočívající v reimplantaci slinných kanálků. V případě CA se však tyto metody považují za neúčinné a otázka zůstává otevřená. Rodiče a ti, kteří se o takovéto pacienty starají, musíme tomuto okamžiku věnovat zvláštní pozornost, protože samotní pacienti obvykle nemají kontrolu nad slinováním a někteří se prostě nemohou o sebe postarat.
Dalším problémem je krátké trvání spánku. Často děti s Angelmanovým syndromem spí ne více než 5 hodin, což negativně ovlivňuje práci celého organismu. Vzrušující, aktivní děti, milující hry a komunikace (i když se snaží omezit se na neverbální způsoby) jsou pro dnešek znatelně unavené. Abyste si odpočinuli dobře, tělo potřebuje plný spánek, ale to je problém.
Mohlo by se zdát, aby zlepšení spánku v drážditelných pacientů by mělo být dostatek léky s sedativní účinky (fenothiaziny a atypických antipsychotik), zklidňuje nervový systém. Ale v případě CA je používání takových léků plné výskytu negativních účinků. Proto lékaři přednost ještě lehké hypnotické drogy, jako je například „Melatonin“ (přírodní hormonální přípravku na základě spánku hormonu), které dávají pacientům hodinu před ulehnutím na 1 tabletu, a „difenhydramin“. Jejichž frekvence podávání a dávkování stanoví lékař, v závislosti na stavu a stáří pacienta.
Někdy pacienti s angelmanovým syndromem mají problémy s trávením a stolicí. Nastavení židle je možné pomocí laxativních přípravků (je to lepší než fytogeneze).
A můžete přistupovat k problému různě, stejně jako američtí lékaři, na základě některé z metod léčby autismu, protože mnoho příznaků charakteristických pro SA, jsou také charakteristické pro autismus (vznětlivost, mimovolné pohyby, opakovaných akcí, deficity pozornosti, problémy v komunikaci, atd. .). Bylo pozorováno, že podávání hormonu secretin, normalizovat zažívání a židle, pozitivní vliv na pozornost pacientů a oxytocin pomáhá zlepšovat kognitivní schopnosti dětí a paměť, opravit chování.
Je pravda, že některé hormony jsou zde nepostradatelné, zejména pokud jde o děti. Angelmanův syndrom ukazuje behaviorální terapii, pracuje s psychologem a řečovým terapeutem (vyučuje neverbální způsoby komunikace a znakový jazyk). Výcvik těchto dětí by měl být založen na individuálním programu za účasti speciálně vyškolených učitelů, psychologů a rodičů. Bohužel to není možné všude a rodiny zůstávají s problémem samy.
Vzhledem k tomu, že mnoho malých pacientů s CA trpí nízkým svalovým tonusem a problémy se spáry, je věnována velká pozornost fyzioterapeutické léčbě. Nejčastěji se lékaři uchylují k použití parafinových aplikací, elektroherose, magnetoterapie.
Aktivní toningová masáž a speciální cvičení fyzioterapie pomohou nemocnému dítěti po chvíli zůstat s důvěrou na nohou a chodit. Zvláště užitečná v tomto ohledu je aquagymnastika, která se doporučuje v CA v chladné vodě. Zvyšuje tón svalů a učí dítě, aby vlastnil své tělo, koordinoval pohyby.
Antikonvulsivní léčba
Nejnebezpečnějším příznakem u Anghelmanova syndromu jsou záchvaty podobné epileptickým záchvatům. Tento příznak je pozorován u 80% pacientů, což znamená, že všem je třeba předepsat účinnou antikonvulzivní léčbu.
Léčba epileptických záchvatů se provádí pomocí vitaminů a antikonvulziv. Při Angelmanův syndrom, doprovázené křečovitý syndrom, bude užitečné vitamíny skupiny B, jakož i vitaminy C, D a E. Ale vitamín terapie jmenovat jejich vlastní v tomto případě je velmi nebezpečné, protože nekontrolované příjem vitaminů může snížit účinnost antiepileptik a vyvolat nový, závažnější a delší záchvaty.
Výběr antikonvulzivních léků a stanovení jejich účinné dávky by měl rovněž provádět odborný lékař. On také rozhodne, zda bude stačit jedna droga, nebo pacient bude muset užívat 2 nebo více léků po dlouhou dobu .
Většina pacientů lékaři předepisují drogy kyselinu valproovou ( „kyselinu valproovou“, „“, „Depakinum Konvuleks“, „valparin“ et al.), Které zabraňují vzniku křečí, zlepšit náladu a duševní stav pacientů.
Kyselina valproová je dostupná ve formě tablet, sirupu a injekčních roztoků. Nejoblíbenějším léčivým přípravkem je droga s prodlouženým účinkem "Depakin" v tabletách a jako roztok pro intravenózní podání. Dávka léčiva je stanovena lékařem individuálně v závislosti na hmotnosti, věku a stavu pacienta.
Vezměte drogu během jídla 2 až 3 krát denně. Průměrná denní dávka je 20-30 mg na 1 kilogram hmotnosti pacienta, maximální je 50 mg / kg denně.
Kontraindikace. Nepoužívá se při porušení jater a pankreatu, hemoragické diatézy, hepatitidy, porfyrie a přecitlivělosti na léčivo.
Mezi nežádoucí účinky lze rozlišovat třesení ruky, trávení a stolici, změny tělesné hmotnosti.
"Topiramát" je také lékem volby v CA. Je vyrobena ve formě tablet a používá se jako součást monoterapie a v kombinaci s jinými léky.
Způsob aplikace a dávkování. Vezměte pilulky uvnitř bez ohledu na příjem potravy. Počáteční denní příjem pro dospělé je 25-50 mg, pro děti 0,5-1 mg / kg. Každý týden se dávka zvyšuje podle lékařského předpisu.
Lék by neměl být užíván během těhotenství a laktace, ale také se zvýšenou citlivostí na jeho složky. Lék má mnoho různých vedlejších účinků.
Léky, které může lékař předepsat na Angelmanovým syndromem "Klomazepam", "", "Rivotril Lamotrigin", "Seyzar", "", "Lamictal levetiracetamu", "Keppra", "Epiterra" et al.
Alternativní léčba a homeopatie
Alternativní medicína, jako homeopatické léky, určitě liší komparativní bezpečnost, avšak účinnost této léčby ve vztahu k Angelholmově syndromu může být považována za spornou.
I když některé alternativní léčby mohou stále pomoci. Jedná se o zastavení epileptických záchvatů. V tomto ohledu může být bylinná terapie docela efektivní.
Dobrým účinkem je lékařský poplatek založený na pivoně, sladovce a kachnu (složky jsou odebírány ve stejném množství). Trávy je třeba mleté na mouku. Po 2 týdnech od začátku příjmu můžete zaznamenat významný pokles frekvence křečových záchvatů.
Užitečné pro křeče a odvar z levandule (1 lžička na sklenici vroucí vody). Formulát se vaří po dobu 5 minut a trval na půl hodiny. Lék užívejte přes noc po dobu 14 dnů.
Efektivní pro epileptické záchvaty je považována za vodní (nebo alkoholovou) infuzi motherwort.
Od homeopatie léky pro prevenci záchvatů s Angelmanův syndrom lze použít léky založené na heřmánek a motherwort, Kyselina hydrocyanicum, Argentum nitricum, draslíku Bromatum, Arsenicum alba. Je však třeba zvážit, že účinné a bezpečné dávky přípravků v každém konkrétním případě mohou jmenovat pouze doktora homeopatika.
Prevence
Jak čtenář pravděpodobně již pochopil, aby se zabránilo mutaci genů a dalších chromozomálních abnormalit, lék je stále nad rámec moci, stejně jako k nápravě situace. To se může stát všem, protože děti se syndromem Angelmanu se narodily také u zdravých rodičů a genetika, která je v současné době jednou z nejméně studovaných oborů medicíny, to zatím neumí vysvětlit.
Jediné, co lze udělat, je převzít zodpovědnost v souvislosti s plánováním těhotenství, aby bylo možné je zaregistrovat a prověřit včas. Opět, takové opatření by však nebylo profylaktické, nýbrž kognitivní, jako každý průzkum. Ale mladí rodiče předem vědí, na co se mají připravit, a v případě pozitivní odpovědi se rozhodnou, zda budou schopni převzít odpovědnost za získání nemocného dítěte.
Předpověď
Prognóza Anghelmanova syndromu závisí na povaze chromozomální abnormality a včasnosti jeho detekce. Nejtěžší je pro ty děti, jejichž 15 chromozomů obsahuje "chybějící" geny (vypouští se). Pravděpodobnost chůze a mluvení u takových pacientů je velmi malá. Zbývající případy s pozorným přístupem a láskou k vašemu dítěti jsou přizpůsobitelné.
Tito pacienti, bohužel, se nemohou stát plnoprávnými členy společnosti, přestože jsou zdaleka hloupí, rozumí řeči a jejím významu. Zde jsou jen problémy s komunikací, kterou mají pro život. Pacienti mohou být od dětství vyučováni znakovou řečí, ale člověk nemůže být nucen komunikovat slovy. Slovníček "mluvících" pacientů je omezen na minimum slov používaných v každodenním životě (5-15 slov).
Pokud jde o očekávanou délku života a celkové zdraví pacientů s Anghelmanovým syndromem, čísla zde kolísají v průměru. V dospělosti pacienty obvykle čelí zdravotním potížím, jako je skolióza a obezita, které s vhodným přístupem k léčbě nejsou život ohrožující.