Lékařský expert článku

Nové publikace
Martin-Bellův syndrom
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
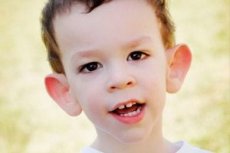
Martinův-Bellův syndrom byl popsán v roce 1943 lékaři, po nichž byl pojmenován. Jedná se o genetickou poruchu spočívající v mentální retardaci. V roce 1969 byly identifikovány změny na chromozomu X (fragilita v distálním rameni) charakteristické pro toto onemocnění. V roce 1991 vědci objevili gen zodpovědný za rozvoj tohoto onemocnění. Toto onemocnění se také nazývá „syndrom fragilního X“. K tomuto onemocnění jsou náchylní chlapci i dívky, ale chlapci jsou postiženi častěji (3krát).
Epidemiologie
Martin-Bellův syndrom je poměrně časté onemocnění: trpí jím 0,3–1,0 z 1 000 mužů a 0,2–0,6 z 1 000 žen. Děti s Martin-Bellovým syndromem se navíc rodí na všech kontinentech se stejnou frekvencí. Národnost, barva pleti, tvar očí, životní podmínky a blahobyt lidí samozřejmě neovlivňují výskyt onemocnění. Jeho frekvence výskytu je srovnatelná pouze s frekvencí Downova syndromu (1 onemocnění z 600–800 novorozenců). Pětina mužských nositelů pozměněného genu je zdravá, nemá žádné klinické ani genové abnormality, zbytek má známky mentální retardace od mírné až po těžkou formu. Mezi ženskými nositelkami je nemocných o něco více než třetina.
Syndrom fragilního X postihuje přibližně 1 z 2 500–4 000 mužů a 1 ze 7 000–8 000 žen. Prevalence nosičství u žen se odhaduje na 1 ze 130–250; prevalence nosičství u mužů se odhaduje na 1 z 250–800.
Příčiny Martin-Bellův syndrom
Martinův-Bellův syndrom se vyvíjí v důsledku úplného nebo částečného zastavení produkce specifického proteinu v těle. K tomu dochází v důsledku absence odpovědi genu FMR1, lokalizovaného v chromozomu X. Mutace vzniká v důsledku restrukturalizace genu z nestabilních strukturních variant genových stavů (alel), a nikoli od samého začátku. Onemocnění se přenáší pouze po mužské linii a muž nemusí být nutně nemocný. Mužští nositelé předávají gen svým dcerám v nezměněné formě, takže jejich mentální retardace není zřejmá. Při dalším přenosu genu z matky na její děti gen mutuje a objevují se všechny příznaky charakteristické pro toto onemocnění.
Patogeneze
Patogeneze Martin-Bellova syndromu je založena na mutacích genového aparátu, které vedou k blokování produkce proteinu FMR, což je protein životně důležitý pro tělo, zejména v neuronech, a je přítomen v různých tkáních. Výzkum ukazuje, že proteiny FMR se přímo podílejí na procesech regulace translací, které probíhají v mozkové tkáni. Absence tohoto proteinu nebo jeho omezená produkce v těle vede k mentální retardaci.
V patogenezi onemocnění je genová hypermetylace považována za klíčovou poruchu, ale dosud nebylo možné definitivně identifikovat mechanismus vzniku této poruchy.
Současně byla zjištěna i lokusová heterogenita patologie, která je spojena s polyalelismem, a také polylokus. Byla stanovena přítomnost alelických variant vývoje onemocnění, které jsou způsobeny existencí bodových mutací a také destrukcí genu typu FMRL.
Pacienti mají také 2 fragilní triplety citlivé na kyselinu listovou, umístěné 300 kb, a také 1,5-2 miliony bp z fragilního tripletu obsahujícího gen FMR1. Mechanismus mutací vyskytujících se v genech FRAXE a FRAXF (jsou identifikovány ve výše zmíněných fragilních tripletech) souvisí s mechanismem poruch u syndromu Martina Bella. Tento mechanismus je způsoben šířením GCC a CGG repetic, které způsobují metylaci tzv. CpG ostrovů. Kromě klasické formy patologie existují i 2 vzácné typy, které se liší v důsledku expanze trinukleotidových repetic (u mužské a ženské meiózy).
Bylo zjištěno, že u klasické formy syndromu pacientovi chybí speciální nukleocytoplazmatický protein typu FMR1, který plní funkci vazby různých mRNA. Kromě toho tento protein podporuje tvorbu komplexu, který pomáhá provádět translační procesy uvnitř ribozomů.
Symptomy Martin-Bellův syndrom
Jak rozpoznat onemocnění u dětí? Jaké jsou první příznaky? V prvních měsících života dítěte není možné rozpoznat Martinův-Bellův příznak, někdy se pozoruje pokles svalového tonu. Po roce je klinický obraz onemocnění zřetelnější: dítě začíná chodit a mluvit pozdě, někdy řeč zcela chybí. Je hyperaktivní, náhodně mává rukama, bojí se davů a hluku, je tvrdohlavé, objevují se prudké výbuchy hněvu, je emoční nestabilita, objevují se epileptické záchvaty, nenavazuje oční kontakt. U pacientů s Martinovým-Bellovým syndromem se onemocnění projevuje i jejich vzhledem: uši jsou odstávající a velké, čelo je těžké, obličej je protáhlý, brada vyčnívá, strabismus, široké ruce a nohy. Vyznačují se také endokrinními poruchami: často velkou hmotností, obezitou, u mužů velkými varlaty, předčasnou pubertou.

U pacientů s Martin-Bellovým syndromem se úroveň inteligence značně liší: od mírné mentální retardace až po těžké případy. Pokud má normální člověk inteligenční kvocient (IQ) v průměru 100 a geniální člověk 130, pak lidé náchylní k tomuto onemocnění mají 35–70.
Všechny klinické příznaky patologie lze charakterizovat triádou hlavních projevů:
- oligofrenie (IQ je 35-50);
- dysmorfofobie (pozorují se vyčnívající uši a prognatismus);
- makroorchidismus, který se objevuje po nástupu puberty.
Přibližně 80 % pacientů má také prolaps bikuspidální chlopně.
Plná forma syndromu se však projevuje pouze u 60 % všech pacientů. U 10 % je zjištěna pouze mentální retardace a u zbývajících se onemocnění vyvíjí s jinou kombinací symptomů.
Mezi první příznaky onemocnění, které se objevují v raném věku:
- nemocné dítě vykazuje ve srovnání s vývojem ostatních vrstevníků výraznou mentální retardaci;
- poruchy pozornosti a koncentrace;
- silná tvrdohlavost;
- děti začínají chodit a mluvit poměrně pozdě;
- pozoruje se hyperaktivita a poruchy vývoje řeči;
- velmi silné a nekontrolovatelné záchvaty hněvu;
- může se vyvinout mutismus - jedná se o úplný nedostatek řeči u dítěte;
- dítě zažívá sociální úzkost a je schopné panikařit kvůli hlasitému hluku nebo jiným hlasitým zvukům;
- dítě nekontrolovatelně a chaoticky mává rukama;
- je pozorována plachost, dítě se bojí být v přeplněných místech;
- vznik různých obsedantních myšlenek, nestabilní emoční stav;
- Dítě se může zdráhat navazovat oční kontakt s lidmi.
U dospělých se pozorují následující příznaky patologie:
- specifický vzhled: protáhlý obličej s těžkým čelem, velké vyčnívající uši, silně vyčnívající brada;
- ploché nohy, zánět středního ucha a strabismus;
- puberta nastává poměrně brzy;
- může se vyvinout obezita;
- U Martin-Bellova syndromu se poměrně často pozorují srdeční vady;
- u mužů je pozorováno zvětšení varlat;
- klouby kloubů se stávají velmi pohyblivými;
- hmotnost a výška prudce stoupají.
Diagnostika Martin-Bellův syndrom
Pro diagnostiku syndromu Martina Bella je nutné kontaktovat kvalifikovaného genetika. Diagnóza se stanoví po provedení specifických genetických testů, které umožňují identifikovat vadný chromozom.
Testy
V rané fázi onemocnění se používá cytogenetická metoda, při které se pacientovi odebere fragment buněčného materiálu, ke kterému se následně přidá kyselina listová, aby se vyvolaly změny v chromozomech. Po určité době se identifikuje oblast chromozomu, kde dochází k jeho znatelnému ztenčení – to je známkou přítomnosti syndromu fragilního X.
Tento test však není vhodný pro diagnostiku v pozdějších stádiích onemocnění, protože jeho přesnost je snížena rozšířeným užíváním multivitaminů obsahujících kyselinu listovou.
Integrovaná diagnostika Martin-Bellova syndromu je molekulárně genetické vyšetření, které spočívá ve stanovení počtu tzv. trinukleotidových repetic v genu.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentální diagnostika
Vysoce specifickou metodou instrumentální diagnostiky je PCR (polymerázová řetězová reakce), která umožňuje studovat strukturu aminokyselinových zbytků obsažených v chromozomu X a tím určit přítomnost syndromu Martina Bella.
Existuje také samostatná, ještě specifičtější metoda diagnostiky patologie – kombinace PCR a detekce pomocí kapilární elektroforézy. Tato metoda je vysoce přesná a detekuje chromozomální patologii u pacientek s primárním selháním vaječníků a také ataxickým syndromem.
Přítomnost defektu lze zjistit po provedení diagnostiky na EEG. Pacienti s tímto onemocněním mají podobnou bioelektrickou aktivitu mozku.
Jaké testy jsou potřeba?
Diferenciální diagnostika
Mezi diferencované metody, které pomáhají podezřívat syndrom, patří:
- klinické - 97,5 % pacientů má zjevné známky mentální retardace (střední nebo těžké); 62 % má odstávající velké uši; 68,4 % má velkou odstávající bradu a čelo; 68,4 % chlapců má zvětšená varlata, 41,4 % má řečové zvláštnosti (nerovnoměrné tempo řeči, nekontrolovatelná hlasitost atd.);
- cytogenní - krev se vyšetřuje na lymfocytární kulturu, stanoví se počet buněk s fragilním chromozomem X na 100 studovaných buněk;
- Elektroencefalografie - zaznamenávají se změny elektrických impulsů mozku, které jsou specifické pro Martin-Bellův syndrom.
Kdo kontaktovat?
Léčba Martin-Bellův syndrom
Při léčbě dospělých pacientů se používají antidepresiva s psychostimulancii. Proces lékové terapie je neustále sledován psychologem a psychiatrem. Kromě toho se v soukromých klinikách provádějí mikroinjekční procedury s léky, jako je Cerebrolysin (nebo jeho deriváty), a také cytomediny (jako je Solcoseryl nebo Lidase).
Při rozvoji ataxického syndromu se používají léky na ředění krve a nootropika. Dále se předepisují směsi aminokyselin a angioprotektory. Ženám s primárním selháním vaječníků je předepsána korektivní léčba pomocí bylinných léků a estrogenů.
V léčbě se také používají antagonisté glutaminových receptorů.
Tradičně léčba Martin-Bellova syndromu zahrnuje užívání léků, které ovlivňují příznaky onemocnění, ale nikoli jeho příčinu. Tato terapie zahrnuje předepisování antidepresiv, neuroleptik a psychostimulancií. Ne všechny léky jsou indikovány k použití u dětí, takže seznam léků je poměrně omezený. Mezi neuroleptika, která lze použít po 3 letech (nejranější věk pro jejich předepisování), patří haloperidol v kapkách a tabletách, chlorpromazin v roztoku a periciazin v kapkách. Dávka haloperidolu pro děti se tedy vypočítává v závislosti na tělesné hmotnosti. U dospělých se dávka předepisuje individuálně. Užívá se perorálně, počínaje 0,5–5 mg 2–3krát denně, poté se dávka postupně zvyšuje na 10–15 mg. Po zlepšení se přechází na nižší dávku, aby se udržel dosažený stav. V případě psychomotorické agitace se podává 5–10 mg intramuskulárně nebo intravenózně, několik opakování je možné po 30–40 minutách. Denní dávka by neměla překročit 100 mg. Možné jsou nežádoucí účinky ve formě nevolnosti, zvracení, svalových křečí, zvýšeného krevního tlaku, arytmie atd. Starší osoby by měly dbát zvláštních opatrností, protože byly zaznamenány případy náhlé zástavy srdce a může se objevit tardivní dyskineze (mimovolní pohyby).
Antidepresiva zvyšují aktivitu mozkových struktur, zmírňují depresi, napětí a zlepšují náladu. Mezi tyto léky, doporučené pro užívání od 5-8 let u Martin-Bellova syndromu, patří klomipromin, sertralin, fluoxetin a fluvoxamin. Fluoxetin se tedy užívá perorálně během jídla 1-2krát (nejlépe v první polovině dne), počínaje dávkou 20 mg denně, v případě potřeby se zvyšuje na 80 mg. Starším lidem se nedoporučuje dávka vyšší než 60 mg. Průběh léčby určuje lékař, ale ne déle než 5 týdnů.
Možné nežádoucí účinky: závratě, úzkost, tinnitus, ztráta chuti k jídlu, tachykardie, otoky atd. Opatrnost je nutná při předepisování starším osobám, osobám s kardiovaskulárními onemocněními a diabetem.
Psychostimulancia jsou psychotropní léky používané ke zlepšení vnímání vnějších podnětů: zostřují sluch, reakční reakce a zrak.
Diazepam se předepisuje jako sedativum při neurózách, úzkosti, epileptických záchvatech a křečích. Užívá se perorálně, intravenózně, intramuskulárně, rektálně (do konečníku). Předepisuje se individuálně v závislosti na závažnosti onemocnění, s nejmenšími dávkami 5-10 mg, denně - 5-20 mg. Délka léčby je 2-3 měsíce. U dětí se dávka vypočítává s ohledem na tělesnou hmotnost a individuální charakteristiky. Mezi nežádoucí účinky patří letargie, apatie, ospalost, nevolnost, zácpa. Je nebezpečné kombinovat s alkoholem, je možná závislost na droze.
Při léčbě Martin-Bellova syndromu byly hlášeny případy zlepšení stavu a zavedením léků vyrobených ze zvířecího materiálu (mozku): cerebrolysát, cerebrolysin, cerebrolysát-M. Hlavními složkami těchto léků jsou peptidy, které podporují produkci proteinů v neuronech, a tím doplňují chybějící protein. Cerebrolysin se podává injekčně v dávce 5-10 ml, léčebná kúra zahrnuje 20-30 injekcí. Lék se předepisuje dětem od jednoho roku věku, podává se intramuskulárně každý den 1-2 ml po dobu jednoho měsíce. Možné jsou opakované aplikace. Nežádoucí účinky ve formě horečky, kontraindikováno u těhotných žen.
Objevily se pokusy o léčbu onemocnění kyselinou listovou, ale zlepšil se pouze behaviorální aspekt (snížila se úroveň agrese a hyperaktivity, zlepšila se řeč) a na intelektuální úrovni se nic nezměnilo. Pro zlepšení stavu onemocnění se předepisuje kyselina listová, indikují se fyzioterapeutické metody, logopedie, pedagogická a sociální korekce.
Lithiové přípravky jsou také považovány za účinné, protože pomáhají zlepšit adaptaci pacienta na sociální prostředí a také kognitivní aktivitu. Kromě toho také regulují jeho chování ve společnosti.
Použití bylin při syndromu Martin-Bell je možné jako antidepresiva. Mezi byliny, které pomáhají zmírnit napětí, úzkost a zlepšit spánek, patří kozlík lékařský, máta peprná, tymián, třezalka tečkovaná a heřmánek. Nálevy se připravují takto: na 1 čajovou lžičku sušených bylin budete potřebovat sklenici vroucí vody, odvary se louhují alespoň 20 minut a užívají se hlavně večer před spaním nebo odpoledne. Dobrým doplňkem by byla lžička medu.
Fyzioterapeutická léčba
K odstranění neurologických projevů se provádějí speciální fyzioterapeutické postupy - jako jsou cvičení v bazénu, uvolnění svalů a akupunktura.
Chirurgická léčba
Za důležitou fázi léčby se považují také metody plastické chirurgie - operace, které pomáhají zlepšit vzhled pacienta. Provádí se plastická chirurgie končetin a ušních boltců, stejně jako genitálií. Provádí se také korekce gynekomastie s epispadiemi a dalších vad vzhledu.
Prevence
Jedinou metodou prevence onemocnění je prenatální screening těhotných žen. Existují speciální vyšetření, která umožňují včasnou detekci patologie, po kterých se doporučuje těhotenství ukončit. Alternativně se používá IVF, které může dítěti pomoci zdědit zdravý chromozom X.
Prevence pacienta závisí na tom, zda genová mutace vznikla znovu, nebo byla zděděna. Za tímto účelem se provádí molekulárně genetická diagnostika. Skutečnost, že test u příbuzných neodhalil „fragilní chromozom X“, hovoří ve prospěch „čerstvosti“ mutace, což znamená, že riziko narození dítěte se syndromem Martin-Bell je velmi malé. V rodinách, kde jsou nemocní lidé, test pomůže vyhnout se opakovaným případům.
Předpověď
Prognóza Martin-Bellova syndromu je příznivá pro život, ale ne pro uzdravení. Délka života závisí na závažnosti onemocnění a souvisejících vadách. Pacient může žít normálním životem. U těžkých forem Martin-Bellova syndromu jsou pacienti ohroženi celoživotní invaliditou.
Očekávaná délka života
Syndrom Martina Bella nemá vážný negativní dopad na zdraví, takže délka života většiny lidí, u kterých byla tato patologie diagnostikována, se neliší od standardních ukazatelů.