Lékařský expert článku

Nové publikace
Dědičná nefritida (Alportův syndrom) u dětí
Naposledy posuzováno: 05.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
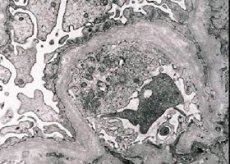
Hereditární nefritida (Alportův syndrom) je geneticky podmíněná dědičná neimunitní glomerulopatie, projevující se hematurií (někdy s proteinurií), progresivním poklesem funkce ledvin s rozvojem chronického selhání ledvin, často kombinovaným se senzorineurální hluchotou a zrakovým postižením.
Toto onemocnění poprvé popsal v roce 1902 LG Guthrie, který pozoroval rodinu, v níž byla hematurie pozorována v několika generacích. V roce 1915 popsal A. F. Hurst rozvoj urémie u členů téže rodiny. V roce 1927 A. Alport poprvé identifikoval ztrátu sluchu u několika příbuzných s hematurií. V 50. letech 20. století byly popsány oční léze u podobného onemocnění. V roce 1972 u pacientů s hereditární hematurií Hinglais a kol. během morfologické studie ledvinové tkáně odhalili nerovnoměrné rozpínání a stratifikaci glomerulárních bazálních membrán. V roce 1985 byl identifikován genetický základ hereditární nefritidy - mutace v genu pro kolagen typu IV (Fiengold a kol., 1985).
Studium genetické povahy onemocnění nám umožnilo dojít k závěru, že rozdíly ve fenotypových projevech hereditární nefritidy (se ztrátou sluchu nebo bez ní) jsou způsobeny stupněm exprese mutantního genu. V současné době jsou tedy všechny klinické varianty považovány za projevy jednoho onemocnění a termín „hereditární nefritida“ je synonymem pro termín „Alportův syndrom“.
Podle epidemiologických studií se dědičná nefritida vyskytuje s frekvencí 17 na 100 000 dětí.
 [
[ Příčiny Alportova syndromu
Genetickým základem onemocnění je mutace v genu řetězce a-5 kolagenu typu IV. Tento typ je univerzální pro bazální membrány ledvin, kochleární aparát, pouzdro čočky, sítnici a rohovku oka, což bylo prokázáno ve studiích s použitím monoklonálních protilátek proti této frakci kolagenu. V poslední době se ukazuje možnost využití DNA sond pro prenatální diagnostiku hereditární nefritidy.
Zdůrazňuje se důležitost testování všech členů rodiny pomocí DNA sond k identifikaci nositelů mutantního genu, což má velký význam při provádění lékařského a genetického poradenství rodin s tímto onemocněním. Až 20 % rodin však nemá příbuzné trpící onemocněním ledvin, což naznačuje vysokou frekvenci spontánních mutací abnormálního genu. Většina pacientů s hereditární nefritidou má ve svých rodinách jedince s onemocněním ledvin, ztrátou sluchu a poruchami zraku; důležitá jsou pokrevní manželství mezi lidmi s jedním nebo více předky, protože v manželství příbuzných jedinců se zvyšuje pravděpodobnost přijetí stejných genů od obou rodičů. Byly stanoveny autozomálně dominantní, autozomálně recesivní a dominantní, X-vázané přenosové cesty.
U dětí se nejčastěji rozlišují tři typy hereditární nefritidy: Alportův syndrom, hereditární nefritida bez ztráty sluchu a familiární benigní hematurie.
Alportův syndrom je dědičná nefritida se sluchovým postižením. Je založena na kombinované poruše struktury kolagenu glomerulární bazální membrány ledvin, ušních a očních struktur. Gen klasického Alportova syndromu se nachází v lokusu 21-22 q dlouhého raménka chromozomu X. Ve většině případů se dědí dominantním způsobem, vázaným na chromozom X. V tomto ohledu je Alportův syndrom závažnější u mužů, protože u žen je funkce mutantního genu kompenzována zdravou alelou druhého, nepoškozeného chromozomu.
Genetickým základem pro rozvoj hereditární nefritidy jsou mutace v genech alfa řetězců kolagenu typu IV. Je známo šest alfa řetězců kolagenu typu IV G: geny řetězců a5 a a6 (Col4A5 a Col4A5) se nacházejí na dlouhém raménku chromozomu X v zóně 21-22q; geny řetězců a3 a a4 (Col4A3 a Col4A4) se nacházejí na 2. chromozomu; geny řetězců a1 a a2 (Col4A1 a Col4A2) se nacházejí na 13. chromozomu.
Ve většině případů (80-85 %) je detekován X-vázaný typ dědičnosti onemocnění, spojený s poškozením genu Col4A5 v důsledku delece, bodových mutací nebo poruch sestřihu. V současné době bylo nalezeno více než 200 mutací genu Col4A5, které jsou zodpovědné za narušení syntézy a5-řetězců kolagenu typu IV. Při tomto typu dědičnosti se onemocnění projevuje u dětí obou pohlaví, ale u chlapců je závažnější.
Mutace v lokusech genů Col4A3 a Col4A4 zodpovědných za syntézu řetězců a3 a a4 kolagenu typu IV se dědí autozomálně. Podle výzkumu je autozomálně dominantní typ dědičnosti pozorován u 16 % případů hereditární nefritidy a autozomálně recesivní typ u 6 % pacientů. Je známo asi 10 variant mutací genů Col4A3 a Col4A4.
Důsledkem mutací je narušení procesů sestavování kolagenu typu IV, což vede k narušení jeho struktury. Kolagen typu IV je jednou z hlavních složek glomerulární bazální membrány, kochleárního aparátu a oční čočky, jejíž patologie bude detekována v klinice hereditární nefritidy.
Kolagen typu IV, který je součástí glomerulární bazální membrány, se skládá převážně ze dvou řetězců a1 (IV) a jednoho řetězce a2 (IV) a obsahuje také řetězce a3, a4, a5. Nejčastěji je u X-vázané dědičnosti mutace genu Col4A5 doprovázena absencí řetězců a3, a4, a5 a a6 ve struktuře kolagenu typu IV a počet řetězců o1 a a2 v glomerulární bazální membráně se zvyšuje. Mechanismus tohoto jevu není jasný, předpokládá se, že příčinou jsou posttranskripční změny v mRNA.
Absence řetězců a3, a4 a a5 ve struktuře kolagenu typu IV glomerulárních bazálních membrán vede v raných stádiích Alportova syndromu k jejich ztenčení a křehkosti, což se klinicky projevuje častěji hematurií (méně často hematurií s proteinurií nebo pouze proteinurií), ztrátou sluchu a lentikonem. Další progrese onemocnění vede v pozdních stádiích onemocnění k ztluštění a zhoršené propustnosti bazálních membrán s proliferací kolagenu typu V a VI v nich, což se projevuje zvýšením proteinurie a snížením funkce ledvin.
Povaha mutace, která je základem hereditární nefritidy, do značné míry určuje její fenotypový projev. V případě delece chromozomu X se současnou mutací genů Col4A5 a Col4A6 zodpovědných za syntézu řetězců a5 a a6 kolagenu typu IV je Alportův syndrom kombinován s leiomyomatózou jícnu a genitálií. Podle výzkumných údajů je v případě mutace genu Col4A5 spojené s delecí zaznamenána větší závažnost patologického procesu, kombinace poškození ledvin s extrarenálními projevy a časný rozvoj chronického selhání ledvin ve srovnání s bodovou mutací tohoto genu.
Morfologicky elektronová mikroskopie odhaluje ztenčení a stratifikaci glomerulárních bazálních membrán (zejména lamina densa) a přítomnost elektronově denzních granulí. Glomerulární léze mohou být u stejného pacienta heterogenní, od minimálních fokálních mezangiálních lézí až po glomerulosklerózu. Glomerulitida u Alportova syndromu je vždy imunonegativní, což ji odlišuje od glomerulonefritidy. Mezi charakteristické rysy patří rozvoj tubulární atrofie, lymfohistiocytární infiltrace a přítomnost „pěnových buněk“ s lipidovými inkluzemi – lipofágů. S postupem onemocnění se odhaluje ztluštění a výrazná destrukce glomerulárních bazálních membrán.
Jsou odhaleny určité změny v imunitním systému. Pacienti s hereditární nefritidou mají sníženou hladinu Ig A a tendenci ke zvýšení koncentrace IgM v krvi, hladina IgG může být v raných stádiích onemocnění zvýšená a v pozdějších stádiích snížená. Zvýšení koncentrace IgM a G je pravděpodobně jakousi kompenzační reakcí v reakci na nedostatek IgA.
Funkční aktivita T-lymfocytárního systému je snížena; je zaznamenán selektivní pokles B-lymfocytů zodpovědných za syntézu Ig A, fagocytární vazba imunity je narušena, zejména v důsledku narušení chemotaxe a intracelulárních trávicích procesů v neutrofilech.
Při vyšetření biopsie ledvin u pacientů s Alportovým syndromem odhalují data elektronové mikroskopie ultrastrukturální změny v glomerulární bazální membráně: ztenčení, narušení struktury a štěpení glomerulárních bazálních membrán se změnou jejich tloušťky a nerovnými konturami. V raných stádiích hereditární nefritidy defekt určuje ztenčení a křehkost glomerulárních bazálních membrán.
Ztenčení glomerulárních membrán je příznivějším znakem a je častější u dívek. Konstantnějším elektronmikroskopickým znakem u hereditární nefritidy je štěpení bazální membrány a závažnost její destrukce koreluje se závažností procesu.
Příznaky Alportova syndromu u dětí
První příznaky Alportova syndromu ve formě izolovaného močového syndromu jsou nejčastěji zjištěny u dětí prvních tří let života. Ve většině případů je onemocnění zjištěno náhodně. Močový syndrom je zjištěn během preventivní prohlídky dítěte, před přijetím do zařízení péče o děti nebo během akutních respiračních infekcí (ARVI). V případě patologie v moči během ARVI. U hereditární nefritidy, na rozdíl od získané glomerulonefritidy, neexistuje latentní období.
V počáteční fázi onemocnění je zdraví dítěte málo ohroženo, charakteristickým rysem je perzistence a rezistenci močového syndromu. Jedním z hlavních příznaků je hematurie různého stupně závažnosti, pozorovaná ve 100 % případů. Zvýšení stupně hematurie je zaznamenáno během nebo po respiračních infekcích, fyzické aktivitě nebo po preventivním očkování. Proteinurie ve většině případů nepřesahuje 1 g/den, na začátku onemocnění může být nestálá, s postupem procesu se proteinurie zvyšuje. Periodicky se v močovém sedimentu může objevit leukocyturie s převahou lymfocytů, což je spojeno s rozvojem intersticiálních změn.
Následně dochází k částečnému zhoršení funkce ledvin, zhoršuje se celkový stav pacienta: objevuje se intoxikace, svalová slabost, arteriální hypotenze, často se objevuje porucha sluchu (zejména u chlapců) a někdy i porucha zraku. Intoxikace se projevuje bledostí, únavou a bolestmi hlavy. V počátečním stádiu onemocnění je ztráta sluchu ve většině případů zjištěna pouze audiografií. Ztráta sluchu u Alportova syndromu se může objevit v různých obdobích dětství, ale nejčastěji je ztráta sluchu diagnostikována ve věku 6-10 let. Ztráta sluchu u dětí začíná vysokými frekvencemi, dosahuje významného stupně ve vzdušném a kostním vedení, přechází ze ztráty sluchu vedoucí zvuk do ztráty sluchu vnímající zvuk. Ztráta sluchu může být jedním z prvních příznaků onemocnění a může předcházet močovému syndromu.
Ve 20 % případů mají pacienti s Alportovým syndromem změny zrakových orgánů. Nejčastěji zjištěnými anomáliemi jsou anomálie čočky: sferofokie, přední, zadní nebo smíšený lentikonus a různé katarakty. V rodinách s Alportovým syndromem je značný výskyt myopie. Řada výzkumníků v těchto rodinách neustále zaznamenává bilaterální perimakulární změny ve formě jasně bělavých nebo nažloutlých granulací v žlutém tělísku. Tento příznak považují za konstantní symptom s vysokou diagnostickou hodnotou u Alportova syndromu. KS Chugh a kol. (1993) v oftalmologické studii zjistili u pacientů s Alportovým syndromem snížení zrakové ostrosti v 66,7 % případů, přední lentikonus v 37,8 %, retinální skvrny v 22,2 %, kataraktu v 20 % a keratokonus v 6,7 %.
U některých dětí s hereditární nefritidou, zejména při rozvoji selhání ledvin, je zaznamenáno významné zpoždění ve fyzickém vývoji. S postupujícím selháním ledvin se rozvíjí arteriální hypertenze. U dětí je častěji zjištěna v dospívání a ve starších věkových skupinách.
Pacienti s hereditární nefritidou se vyznačují přítomností různých (více než 5-7) stigmat pojivové tkáně. Mezi stigmaty pojivové tkáně u pacientů jsou nejčastější hypertelorismus očí, vysoké patro, anomálie skusu, abnormální tvar boltců, zakřivení malíčku na rukou a „mezera v sandálech“ na nohou. Hereditární nefritida se vyznačuje uniformitou stigmat dysmorfogeneze v rámci rodiny a také vysokou četností jejich rozšíření mezi příbuznými probandů, po jejichž linii se onemocnění přenáší.
V časných stádiích onemocnění je zjištěn izolovaný pokles parciálních renálních funkcí: transport aminokyselin, elektrolytů, koncentrační funkce, acidogeneze, pozdější změny ovlivňují funkční stav proximální i distální části nefronu a jsou charakterizovány kombinovanými parciálními poruchami. K poklesu glomerulární filtrace dochází později, častěji v adolescenci. S progresí hereditární nefritidy se rozvíjí anémie.
Dědičná nefritida se tedy vyznačuje postupným průběhem onemocnění: nejprve latentní stadium nebo skryté klinické příznaky, projevující se minimálními změnami močového syndromu, poté dochází k postupné dekompenzaci procesu se snížením funkce ledvin s manifestními klinickými příznaky (intoxikace, astenie, vývojové zpoždění, anémie). Klinické příznaky se obvykle objevují bez ohledu na vrstvení zánětlivé reakce.
Dědičná nefritida se může projevit v různých věkových obdobích, což závisí na působení genu, který je do určité doby v potlačeném stavu.
Klasifikace
Existují tři typy dědičné nefritidy
- Varianta I - klinicky se projevuje jako nefritida s hematurií, ztrátou sluchu a poškozením očí. Průběh nefritidy je progresivní s rozvojem chronického selhání ledvin. Typ dědičnosti je dominantní, vázaný na chromozom X. Morfologicky se odhaluje porušení struktury bazální membrány, její ztenčení a štěpení.
- Varianta II - klinicky se projevuje jako nefritida s hematurií bez ztráty sluchu. Průběh nefritidy je progresivní s rozvojem chronického selhání ledvin. Typ dědičnosti je dominantní, vázaný na chromozom X. Morfologicky je detekováno ztenčení bazální membrány glomerulárních kapilár (zejména laminadensa).
- Možnost III - benigní familiární hematurie. Průběh je příznivý, chronické selhání ledvin se nevyvíjí. Typ dědičnosti je autozomálně dominantní nebo autozomálně recesivní. U autozomálně recesivního typu dědičnosti je u žen zaznamenán závažnější průběh onemocnění.
Diagnóza Alportova syndromu
Navrhují se následující kritéria:
- přítomnost alespoň dvou pacientů s nefropatií v každé rodině;
- hematurie jako hlavní příznak nefropatie u probanda;
- přítomnost ztráty sluchu u alespoň jednoho člena rodiny;
- rozvoj chronického selhání ledvin u jednoho nebo více příbuzných.
V diagnostice různých dědičných a vrozených onemocnění je velký prostor věnován komplexnímu přístupu k vyšetření a především pozornosti věnované údajům získaným při sestavování rodokmenu dítěte. Diagnóza Alportova syndromu je považována za platnou v případech, kdy jsou u pacienta zjištěny 3 ze 4 typických znaků: přítomnost hematurie a chronického selhání ledvin v rodině, přítomnost neurosenzorické ztráty sluchu, patologie zraku u pacienta, detekce známek štěpení glomerulární bazální membrány se změnou její tloušťky a nerovnými konturami při elektronově mikroskopických charakteristikách biopsie.
Vyšetření pacienta by mělo zahrnovat klinické a genetické výzkumné metody; cílené studium anamnézy onemocnění; celkové vyšetření pacienta s ohledem na diagnosticky významná kritéria. Ve stadiu kompenzace lze patologii detekovat pouze zaměřením na takové syndromy, jako je přítomnost dědičné zátěže, hypotenze, mnohočetná stigma dysembryogeneze, změny v močovém syndromu. Ve stadiu dekompenzace se mohou objevit extrarenální příznaky, jako je těžká intoxikace, astenie, opožděný fyzický vývoj, anémie, které se projevují a zhoršují s postupným snižováním funkce ledvin. U většiny pacientů se sníženou funkcí ledvin se pozoruje: snížená acido- a aminogeneze; 50 % pacientů zaznamenává významný pokles sekreční funkce ledvin; omezený rozsah kolísání optické hustoty moči; porucha filtračního rytmu a následně pokles glomerulární filtrace. Stádium chronického selhání ledvin je diagnostikováno, když mají pacienti zvýšenou hladinu močoviny v krevním séru (více než 0,35 g/l) po dobu 3-6 měsíců nebo déle a pokles glomerulární filtrace na 25 % normy.
Diferenciální diagnostika hereditární nefritidy by měla být prováděna především s hematurickou formou získané glomerulonefritidy. Získaná glomerulonefritida má nejčastěji akutní nástup, období 2–3 týdnů po infekci, extrarenální příznaky, včetně hypertenze od prvních dnů (u hereditární nefritidy naopak hypotenze), sníženou glomerulární filtraci na začátku onemocnění, bez poškození parciálních tubulárních funkcí, zatímco u hereditárních jsou přítomny. Získaná glomerulonefritida se vyskytuje s výraznější hematurií a proteinurií, se zvýšenou sedimentací erytrocytů (ESR). Diagnostickou hodnotu mají typické změny glomerulární bazální membrány, charakteristické pro hereditární nefritidu.
Diferenciální diagnostika od dysmetabolické nefropatie se provádí s chronickým selháním ledvin, v rodině se klinicky projevují heterogenní onemocnění ledvin a může se vyskytovat spektrum nefropatií od pyelonefritidy až po urolitiázu. Děti si často stěžují na bolest břicha a periodicky při močení, v moči sediment - oxaláty.
Pokud existuje podezření na hereditární nefritidu, měl by být pacient odeslán na specializované nefrologické oddělení k objasnění diagnózy.
Co je třeba zkoumat?
Jak zkoušet?
Jaké testy jsou potřeba?
Kdo kontaktovat?
Léčba Alportova syndromu
Režim zahrnuje omezení těžké fyzické námahy a pobytu na čerstvém vzduchu. Strava je kompletní, s dostatečným množstvím kompletních bílkovin, tuků a sacharidů s přihlédnutím k funkci ledvin. Velký význam má detekce a léčba chronických ložisek infekce. Používají se následující léky: ATP, kokarboxyláza, pyridoxin (až 50 mg/den), karnitin chlorid. Kúry se podávají 2-3krát ročně. Při hematurii se předepisuje bylinná medicína - kopřiva dvoudomá, šťáva z arónie, řebříček obecný.
V zahraniční i domácí literatuře existují zprávy o léčbě prednisolonem a použití cytostatik. Je však obtížné posoudit jejich účinek.
Při chronickém selhání ledvin se používá hemodialýza a transplantace ledvin.
Neexistují žádné metody specifické (účinné patogenetické) terapie hereditární nefritidy. Veškerá léčebná opatření jsou zaměřena na prevenci a zpomalení poklesu renálních funkcí.
Strava by měla být vyvážená a kalorická, s ohledem na funkční stav ledvin. Při absenci funkčních poruch by dětská strava měla obsahovat dostatek bílkovin, tuků a sacharidů. Při přítomnosti známek dysfunkce ledvin by mělo být množství bílkovin, sacharidů, vápníku a fosforu omezeno, což zpomaluje rozvoj chronického selhání ledvin.
Fyzická aktivita by měla být omezena; dětem se doporučuje vyhýbat se sportu.
Je třeba se vyhnout kontaktu s infekčními pacienty, snížit riziko vzniku akutních respiračních onemocnění. Je nutná sanitace ložisek chronické infekce. Preventivní očkování se u dětí s hereditární nefritidou neprovádí, očkování je možné pouze z epidemiologických indikací.
Hormonální a imunosupresivní terapie u hereditární nefritidy je neúčinná. Existují náznaky určitého pozitivního účinku (snížení proteinurie a zpomalení progrese onemocnění) při dlouhodobém a víceletém užívání cyklosporinu A a ACE inhibitorů.
Při léčbě pacientů se používají léky, které zlepšují metabolismus:
- pyridoxin - 2-3 mg/kg/den ve 3 dávkách po dobu 4 týdnů;
- kokarboxyláza - 50 mg intramuskulárně obden, celkem 10-15 injekcí;
- ATP - 1 ml intramuskulárně obden, 10-15 injekcí;
- vitamín A - 1000 IU/rok/den v 1 dávce po dobu 2 týdnů;
- Vitamín E - 1 mg/kg/den v 1 dávce po dobu 2 týdnů.
Tento typ terapie pomáhá zlepšit celkový stav pacientů, redukovat tubulární dysfunkce a provádí se v kurzech 3krát ročně.
Levamisol lze použít jako imunomodulátor - 2 mg/kg/den 2-3krát týdně s přestávkami mezi dávkami 3-4 dny.
Podle výzkumných údajů má hyperbarická oxygenace pozitivní vliv na závažnost hematurie a renální dysfunkce.
Nejúčinnější metodou léčby hereditární nefritidy je včasná transplantace ledvin. V tomto případě nedochází k relapsu onemocnění v transplantované ledvině; v malém procentu případů (asi 5 %) se může v transplantované ledvině vyvinout nefritida spojená s antigeny proti glomerulární bazální membráně.
Slibným směrem je prenatální diagnostika a genetické inženýrství. Experimenty na zvířatech ukazují vysokou účinnost přenosu normálních genů zodpovědných za syntézu alfa řetězců kolagenu typu IV do ledvinové tkáně, po čemž je pozorována syntéza normálních kolagenových struktur.
Předpověď
Prognóza hereditární nefritidy je vždy vážná.
Prognosticky nepříznivá kritéria pro průběh hereditární nefritidy jsou:
- mužské pohlaví;
- časný rozvoj chronického selhání ledvin u rodinných příslušníků;
- proteinurie (více než 1 g/den);
- ztluštění glomerulárních bazálních membrán dle mikroskopie;
- akustická neuritida;
- delece v genu Col4A5.
Prognóza benigní familiární hematurie je příznivější.
Использованная литература