Lékařský expert článku

Nové publikace
Alkaptonurie je vrozená enzymová abnormalita.
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Alkaptonurie, jedna z velmi vzácných metabolických poruch, označuje vrozené anomálie v metabolismu aminokyseliny tyrosinu.
Tento syndrom může být také nazýván deficitem homogentisátoxidázy, homogentisinurií, hereditární ochronózou nebo onemocněním černé moči.[ 1 ]
Epidemiologie
Podle statistik se alkaptonurie vyskytuje maximálně devět případů na 1 milion obyvatel. A ve většině evropských zemí je to jeden případ na 100–250 tisíc živě narozených dětí.
Mezi evropskými zeměmi je výjimkou Slovensko (zejména relativně malý severozápadní region), kde je prevalence alkaptonurie jeden případ na 19 000 novorozenců. To je s největší pravděpodobností způsobeno tím, že mezi slovenskými romskými rodinami, které tam žijí, je míra inbreedingu (sňatků mezi bratranci a sestřenicemi) nejvyšší v Evropě: 10–14 %. [ 2 ]
Příčiny alkaptonurie
Přesné příčiny alkaptonurie, jakožto vrozené poruchy katabolismu (metabolického rozkladu) aromatické (homocyklické) α-aminokyseliny tyrosinu, byly stanoveny: tento typ metabolické poruchy je důsledkem homozygotních nebo složených heterozygotních mutací jednoho z tisíců genů na chromozomu 3, přesněji genu HGD v lokusu 3q21-q23 na dlouhém rameni chromozomu. Tento gen kóduje nukleotidové sekvence jaterního enzymu homogentisát-1,2-dioxygenázy [ 3 ] (nazývané také oxidáza kyseliny homogentisové nebo homogentisátoxidáza) – metaloproteinu obsahujícího železo, který je nezbytný pro jednu z fází rozkladu tyrosinu v těle. [ 4 ], [ 5 ]
Alkaptonurie je tedy defekt enzymu homogentisát-1,2-dioxygenázy, nebo přesněji řečeno, důsledek jeho geneticky podmíněného deficitu nebo úplné absence. [ 6 ]
Alkaptonurie, která je vrozeným deficitem enzymu, se dědí autozomálně recesivně, to znamená, že aby se alkaptonurie u dětí vyskytla, musí mít oba rodiče modifikovaný gen pro tento enzym, protože každý z nich předá dítěti pouze jednu kopii genu ze dvou dostupných.
Podle nejnovějších údajů existuje více než dvě stě variant modifikace genu HGD, přičemž nejčastěji se pozorují missense mutace, translokace a splicing.
Rizikové faktory
Jediným rizikovým faktorem pro rozvoj této vrozené enzymopatie je její přítomnost v rodinné anamnéze a dědičnost dvou modifikovaných kopií genu HGD, pokud rodiče nevykazují alkaptonurii (riziko přenosu anomálie je 25 %), nebo pokud jeden z rodičů má tuto poruchu. [ 7 ]
Patogeneze
Tyrosin hraje klíčovou roli v syntéze bílkovin, produkci chromoproteinů – kožního pigmentu melaninu, a také hormonů štítné žlázy a katecholaminových neurotransmiterů.
Mechanismus regulace množství tyrosinu v buňkách je velmi složitý a tělo normalizuje jeho nadbytečný obsah jeho odbouráváním. Proces katabolismu tyrosinu, stejně jako u všech aromatických aminokyselin, je vícestupňový a probíhá v několika fázích. Každá fáze metabolického odbourávání tyrosinu probíhá za účasti specifického enzymu a za vzniku meziproduktu.
Takže nejprve se aminokyselina rozloží na para-hydroxyfenylpyruvát, který se přemění na alkapton – 2,5-dihydroxyfenyloctovou neboli kyselinu homogentisovou. Poté by se alkapton měl přeměnit na kyselinu maleoctovou, ale to se neděje. [ 8 ]
A patogeneze alkaptonurie spočívá v zastavení biochemických reakcí katabolismu tyrosinu ve fázi tvorby kyseliny homogentisové: k jejímu rozkladu prostě není potřeba žádný enzym – homogentisátoxidáza.
Kyselina homogentisová není tělem využívána a může se hromadit vylučováním ledvinami. Kromě toho se oxiduje na benzochinoacetát (kyselinu benzochinonooctovou), který vazbou na molekuly tkání a tělních tekutin vytváří biopolymerní sloučeniny zbarvené jako melanin.
Akumulace těchto meziproduktů v tkáni vede k narušení kolagenní struktury chrupavčité tkáně, což snižuje její elasticitu – s výskytem mnoha klinických příznaků alkaptonurie a rozvojem komplikací.
Symptomy alkaptonurie
Alkaptonurie u novorozenců a kojenců se vyznačuje ztmavnutím moči. Moč na plenkách, plenkách a spodním prádle se při styku se vzduchem zbarví do tmavě hněda; to je způsobeno hromaděním a uvolňováním kyseliny homogentisové, která se oxiduje na benzochinoacetát. [ 9 ]
Při absenci dalších příznaků není alkaptonurie u malých dětí často včas rozpoznána, protože moč může po několika hodinách močení ztmavnout. Podle některých údajů je v klinickém prostředí identifikována pouze pětina dětí mladších 12 měsíců, které se narodily s deficitem tohoto enzymu. Proto je velmi důležité, aby rodiče věnovali péči o své kojence pozornost.
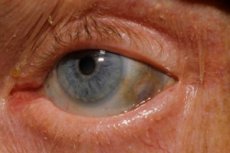
Mezi časné příznaky patří navíc pigmentace (modravě šedá barva) bělimy očí a chrupavek uší a nosu, která se často nazývá ochronóza.[ 10 ]
Postupem času se objevují další příznaky:
- silná pigmentace kůže na lícních kostech, v podpaží a na genitáliích;
- zabarvení oděvu při kontaktu s pocenými oblastmi těla;
- záchvaty celkové slabosti;
- chraplavý hlas.
Je třeba mít na paměti, že alkaptonurie a ochronóza, jak je uvedeno výše, jsou synonyma pro stejnou poruchu katabolismu tyrosinu.
Javorový sirup - onemocnění moči a alkaptonurie. Vrozené javorový sirup - onemocnění moči nebo leucinóza je také metabolická porucha, má stejný vzorec dědičnosti a dokonce i mutace se vyskytují na stejném chromozomu, ale ovlivňují gen kódující enzymový komplex rozvětvené α-ketokyseliny dehydrogenázy. Kvůli tomu tělo nemůže rozkládat určité složky bílkovin, zejména aminokyseliny leucin, isoleucin a valin. U tohoto onemocnění má moč (a ušní maz) sladkou vůni; klinický obraz tohoto typu organické acidémie navíc zahrnuje hypopigmentaci, kolísání krevního tlaku, záchvaty, zvracení a průjem, pokles hladiny glukózy v krvi, ketoacidózu, halucinace atd. Úmrtnost u dětí je poměrně vysoká; u dospělých může bez léčby dojít ke kómatu a smrti v důsledku mozkového edému.
Albinismus a alkaptonurie „spojuje“ pouze tyrosin. Albinismus, včetně okulokutánního, je způsoben genetickými mutacemi, které ovlivňují produkci pigmentu melaninu. Vrozené změny jsou zaznamenány v genu TYR na chromozomu 11 (11q14.3), který kóduje tyrosinázu, enzym melanosomu obsahující měď, nezbytný pro tvorbu kožního pigmentu na bázi produktů metabolismu tyrosinu. Toto onemocnění je mnohem častější než alkaptonurie.
Komplikace a důsledky
Důsledky a komplikace alkaptonurie, způsobené působením meziproduktů tyrosinu – kyseliny homogentisové a benzochinonoctové – se objevují v důsledku ukládání reaktivních pigmentovaných polymerů, destrukce kolagenních fibril a zhoršení stavu chrupavky (se snížením jejich odolnosti vůči mechanickému namáhání).
V průběhu let, v dospělosti, se rozvíjí degenerativní artritida a osteoartróza velkých kloubů (kyčelních, sakroiliakálních a kolenních); meziobratlové prostory se zužují (zejména v bederní a hrudní páteři) – s kalcifikací a tvorbou osteofytů; hustota tkáně subchondrálních kostních plotének se snižuje a podkladové kosti mohou procházet patologickou remodelací s tvorbou výrůstků a deformací. [ 11 ]
V důsledku stejné kalcifikace může být pozorováno poškození srdečních chlopní (aortální a mitrální) a koronárních tepen – s příznaky ischemické choroby srdeční, stejně jako tvorba kamenů v ledvinách a prostatě. [ 12 ], [ 13 ]
Diagnostika alkaptonurie
Diagnóza vrozených metabolických poruch je obvykle založena na studiu biologických tekutin těla.
Na základě jakých testů a reakcí lze diagnostikovat alkaptonurii? K detekci kyseliny homogentisové a stanovení její hladiny (normální – 20–30 mg denně, zvýšená – 3–8 g) jsou nutné testy moči. Vzorek moči se vyšetřuje plynovou chromatografií nebo hmotnostní spektrometrií s použitím kapalinové chromatografie; možný je i screeningový test na přítomnost chloridu železa v moči. [ 14 ]
Existuje také metoda pro rychlou diagnostiku – stanovení alkaptonu v zaschlých skvrnách moči na papíře (intenzitou barvy).
Při objasňování diagnózy zahrnuje instrumentální diagnostika (rentgenografie) identifikaci radiologických příznaků osteoartrózy a dalších kloubních patologií u pacientů.
Diagnóza se potvrzuje molekulárně genetickými metodami diagnostiky dědičných onemocnění, jako je genetické testování a sekvenování DNA. [ 15 ]
Diferenciální diagnostika
Diferenciální diagnóza zahrnuje hemochromatózu a akutní selhání jater novorozence, melaninurii, akutní intermitentní porfyrii, hemofagocytární lymfohistiocytózu, primární mitochondriální patologii, revmatoidní artritidu, ankylozující spondylitidu.
Kdo kontaktovat?
Léčba alkaptonurie
Hlavní léčbou alkaptonurie je perorální podávání velkých dávek (alespoň 1000 mg denně) kyseliny askorbové. U dětí to zvyšuje vylučování kyseliny homogentisové močí a u dospělých to snižuje obsah jejího derivátu, kyseliny benzochinonoctové, v moči a zpomaluje její vazbu na struktury pojivové tkáně kloubů a kolagenu. [ 16 ]
Západoevropské kliniky testují lék Nitisinone (Orfalin), lék ze skupiny metabolitů, který inhibuje druhou fázi katabolismu tyrosinu: transformaci para-hydroxyfenylpyruvátu na kyselinu homogentisovou. Použití tohoto farmakologického činidla však vede k akumulaci tyrosinu a může způsobit závažné vedlejší účinky, včetně zákalu rohovky a fotofobie, krvácení z nosu a žaludku, selhání jater, změn v krvi atd. Nicméně ve Spojených státech je Nitisinone schválen Úřadem pro kontrolu potravin a léčiv (FDA) pro léčbu tyrosinemie typu I. [17 ], [ 18 ]
Proto se u problémů s klouby způsobených alkaptonurií provádí fyzioterapie – cvičení pro zvýšení svalové síly a zlepšení pohyblivosti kloubů, balneoterapie a peloidní terapie pro omezení bolesti.
Přestože tyrosin není dodáván pouze potravou, ale tělo si ho i vytváří, pacientům s alkaptonurií se doporučuje dodržovat dietu s nízkým obsahem bílkovin a omezit konzumaci potravin bohatých na tyrosin, především hovězího a vepřového masa, mléčných výrobků (zejména sýrů), luštěnin, ořechů a semen.
Prevence
Prevence genových mutací je nemožná, ale pro zabránění narození dětí s vysokým rizikem vrozených vad existuje lékařské genetické poradenství, které je nezbytné před plánovaným těhotenstvím u párů, jejichž rodinná anamnéza zahrnuje dědičná onemocnění. [ 19 ]
Předpověď
Smrtelné následky alkaptonurie jsou velmi vzácné a smrt může být způsobena závažnými komplikacemi postihujícími srdce a ledviny. Celková délka života lidí s alkaptonurií je tedy dobrá.
Kvalita života je však snížena v důsledku intenzivní bolesti v kloubech nebo páteři s výrazným omezením pohyblivosti, často progresivní.