Lékařský expert článku

Nové publikace
Achondroplazie
Last reviewed: 12.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
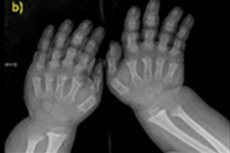
Existuje mnoho vzácných vrozených onemocnění a jedním z nich je porušení růstu kostí - achondroplazie, která vede k těžkému nepřiměřenému nízkému vzrůstu.
V části o vývojových anomáliích MKN-10 je pro tento typ hereditární osteochondrální dysplazie s růstovými vadami tubulárních kostí a páteře použit kód Q77.4 [ 1 ].
Epidemiologie
Pokud jde o prevalenci achondroplazie, statistické údaje z různých studií jsou nejednoznačné. Někteří tvrdí, že se tato anomálie vyskytuje u jednoho novorozence z 10 tisíc, jiní u jednoho z 26–28 tisíc a další u 4–15 případů ze 100 tisíc. [ 2 ]
Existují také informace, že pokud je otec starší 50 let, je výskyt achondroplazie u dětí jeden případ na 1875 novorozenců.
Příčiny achondroplazie
Příčinou achondroplazie je porušení osteogeneze, zejména jeden z typů intrauterinní osifikace diafýz tubulárních kostí kostry - endochondrální osifikace, během níž se chrupavka modifikuje na kostní tkáň. Více informací viz - Vývoj a růst kostí
K narušení osifikace dlouhých kostí, tj. fetální achondroplazie, dochází v důsledku mutací v genu membránové tyrozinkinázy - receptoru 3 fibroblastového růstového faktoru (FGFR3 na chromozomu 4p16.3), který ovlivňuje růst a diferenciaci buněk. Přítomnost mutací FGFR3 je spojena s genetickou nestabilitou a změnami v počtu chromozomů (aneuploidie).
Achondroplazie se na dítě přenáší jako autozomálně dominantní znak, to znamená, že na páru nepohlavních (autozomálních) chromozomů obdrží jednu kopii mutantního genu (který je dominantní) a jeden normální gen. Typ dědičnosti této vady je tedy autozomálně dominantní a anomálie se může projevit u 50 % potomků při křížení kombinace alel tohoto genu (genotypu).
Kromě toho mohou být mutace sporadické a, jak ukazuje praxe, v 80 % případů se děti s achondroplazií rodí rodičům normální výšky.
Rizikové faktory
Hlavními rizikovými faktory pro narození dětí s achondroplazií jsou dědičné. Pokud má jeden z rodičů tuto vadu, pak se pravděpodobnost narození nemocného dítěte odhaduje na 50 %; pokud mají tuto anomálii oba rodiče, je to také 50 %, ale s 25% rizikem homozygotní achondroplazie, vedoucí k úmrtí před narozením nebo v raném kojeneckém věku.
S věkem otce (blíže 40 letům a více) se zvyšuje riziko nové mutace (de novo mutace) genu FGFR3.
Patogeneze
Při vysvětlování patogeneze achondroplazie odborníci zdůrazňují význam transmembránové proteinové tyrosinové proteinkinázy (kódované genem FGFR3) v regulaci dělení, diferenciace a apoptózy buněk chrupavčité tkáně růstových plotének - chondrocytů, a také normálního vývoje kostry - osteogeneze a mineralizace kostní tkáně.
Během embryonálního vývoje, v přítomnosti genové mutace, se receptory fibroblastového růstového faktoru 3 aktivují. Zvýšení jejich funkcí narušuje přenos buněčných signálů a interakci extracelulární části tohoto proteinu s polypeptidovými fibroblastovými růstovými faktory (FGF). V důsledku toho dochází k selhání: fáze proliferace chrupavčitých buněk se zkracuje a jejich diferenciace začíná dříve, než se očekávalo. To vše vede k nesprávné tvorbě a srůstu lebečních kostí a skeletální dysplazii - úbytku dlouhých kostí, který je doprovázen výrazným nízkým vzrůstem neboli nanismem.
A dvě třetiny případů nanismu jsou spojeny s achondroplazií.
Symptomy achondroplazie
Abnormální růst kostí způsobuje klinické příznaky achondroplazie, jako například:
- výrazný nízký vzrůst (neproporcionální nanismus) s průměrnou výškou v dospělosti 123-134 cm;
- zkrácení proximálních částí dolních a horních končetin při relativně normální velikosti trupu;
- zkrácené prsty na rukou a nohou;
- zvětšená hlava (makro- nebo megalocefalie); [ 3 ]
- specifické rysy obličeje v podobě vystouplého čela a hypoplazie střední části obličeje - propadlý nosní hřbet.
- úzký kraniocervikální přechod. Někteří kojenci s achondroplazií umírají v prvním roce života na komplikace související s kraniocervikálním přechodem; populační studie naznačují, že toto zvýšené riziko úmrtí může bez vyšetření a intervence dosahovat až 7,5 %.[ 4 ]
- Dysfunkce středního ucha je často problémem [ 5 ] a pokud není správně léčena, může vést k převodní ztrátě sluchu, která je natolik závažná, že narušuje vývoj řeči. Více než polovina dětí bude potřebovat trubici pro vyrovnání tlaku. [ 6 ] Celkově má asi 40 % lidí s achondroplazií funkčně významnou ztrátu sluchu. Vývoj expresivní řeči je také často zpožděn, ačkoli síla vztahu mezi ztrátou sluchu a problémy s expresivní řečí je sporná.
- Prohnutí holenní kosti je u lidí s achondroplazií velmi časté. Více než 90 % neléčených dospělých má určitý stupeň prohnutí.[ 7 ] „Prohnutí“ je ve skutečnosti komplexní deformita vyplývající z kombinace laterálního naklonění, vnitřní torze holenní kosti a dynamické nestability kolena.[ 8 ]
Kojenci s achondroplazií se vyznačují svalovou hypotonií, kvůli které se začínají později učit pohybovým dovednostem a chodit. Inteligence a kognitivní schopnosti nejsou touto vývojovou vadou ovlivněny. [ 9 ], [ 10 ]
Důsledky a komplikace
Tento typ dědičné osteochondrální dysplazie je charakterizován následujícími komplikacemi a následky:
- opakované infekce ucha;
- obstrukční spánková apnoe;
- hydrocefalus;
- malokluze a křivé zuby:
- deformace nohou (varus nebo valgus) se změnou chůze;
- hypertrofická lordóza bederní páteře nebo její zakřivení (torakolumbální kyfóza nebo bederní skolióza) - s bolestmi zad při chůzi;
- bolest kloubů (v důsledku nesprávného umístění kostí nebo komprese nervových kořenů);
- Spinální stenóza a komprese míchy; Nejčastějším zdravotním problémem v dospělosti je symptomatická spinální stenóza postihující L1-L4. Příznaky sahají od intermitentní, reverzibilní klaudikace vyvolané cvičením až po těžkou, nevratnou dysfunkci nohou a retenci moči.[ 11 ] Klaudikace a stenóza mohou způsobovat jak senzorické (necitlivost, bolest, tíha), tak motorické příznaky (slabost, klopýtaní, omezená vytrvalost při chůzi). Cévní klaudikace je důsledkem otoku cév po postavení se a chůzi a je zcela reverzibilní v klidu. Spinální stenóza je skutečná léze míchy nebo nervového kořene stenotickou kostí páteřního kanálu a příznaky jsou nevratné. Příznaky lokalizované v konkrétním dermatomu mohou být důsledkem stenózy specifických foram nervového kořene.
- zmenšení hrudní stěny s omezeným růstem plic a sníženou plicní funkcí (těžká dušnost). V kojeneckém věku má malá skupina lidí s achondroplazií restriktivní plicní problémy. Malá prsa a zvýšená poddajnost hrudníku v kombinaci vedou ke snížené kapacitě plic a restriktivnímu plicnímu onemocnění [ 12 ]
Další ortopedické problémy
- Slabost kloubů. Většina kloubů je v dětství hypermobilní. Obecně to má malý vliv, s výjimkou nestability kolene u některých lidí.
- Diskoidní laterální meniskus: Tato nedávno identifikovaná strukturální abnormalita může u některých lidí vést k chronické bolesti kolene.[ 13 ]
- Artritida: Konstitutivní aktivace FGFR-3, jako u achondroplazie, může chránit před rozvojem artritidy.[ 14 ]
- Acanthosis nigricans se vyskytuje přibližně u 10 % lidí s achondroplazií.[ 15 ] V této populaci neodráží hyperinzulinémii ani maligní onemocnění.
Homozygotní achondroplazie způsobená bialelickými patogenními variantami na nukleotidu 1138 genu FGFR3 je závažné onemocnění s radiologickými nálezy kvalitativně odlišnými od nálezů pozorovaných u achondroplazie. Předčasná smrt je důsledkem respiračního selhání v důsledku malé hrudní stěny a neurologických deficitů v důsledku cervikomedulární stenózy [Hall 1988].
Diagnostika achondroplazie
U většiny pacientů je diagnóza achondroplazie stanovena na základě charakteristických klinických příznaků a rentgenových nálezů. U kojenců nebo při absenci některých příznaků se k definitivní diagnóze používá genetické testování, jako je analýza karyotypu.[ 16 ]
Při provádění prenatální diagnostiky metodou molekulární genetiky lze provést analýzy plodové vody nebo vzorku choriových klků.
Známky achondroplazie na ultrazvuku plodu - zkrácení končetin a typické rysy obličeje - jsou vizualizovány po 22 týdnech těhotenství.
Instrumentální diagnostika zahrnuje také rentgen kostry nebo ultrazvuk kostí. Rentgen potvrzuje diagnózu na základě údajů, jako je velká lebka s úzkým týlním otvorem a relativně malou bází; krátké trubkovité kosti a zkrácená žebra; krátká a zploštělá těla obratlů; zúžený páteřní kanál, zmenšená velikost křídel kyčelních kostí.
Diferenciální diagnostika
Diferenciální diagnostika s hypofyzárním nanismem, vrozenou spondyloepifyzeální a diastrofickou dysplazií, hypochondroplazií, Shereshevsky-Turnerovým a Noonanovým syndromem a pseudoachondroplazií je nutná. Rozdíl mezi pseudoachondroplazií a achondroplazií tedy spočívá v tom, že u pacientů s nanismem při pseudoachondroplazii je velikost hlavy a rysy obličeje normální.
Kdo kontaktovat?
Léčba achondroplazie
Doporučení pro péči o děti s achondroplazií byla nastíněna Výborem pro genetiku Americké akademie pediatrie. Tato doporučení slouží jako vodítko a nenahrazují individuální rozhodování. Nedávný přehled [Pauli & Botto 2020] také obsahuje pokyny. Existují specializované kliniky, které se specializují na léčbu skeletální dysplazie; jejich doporučení se mohou mírně lišit od těchto obecných doporučení.
Doporučení zahrnují (mimo jiné) následující.
Hydrocefalus. Pokud se objeví známky nebo příznaky zvýšeného nitrolebního tlaku (např. zrychlený růst hlavy, přetrvávající vypouklá fontanela, znatelné zvětšení povrchových žil na obličeji, podrážděnost, zvracení, změny vidění, bolest hlavy), je nutné odeslat pacienta k neurochirurgovi.
Předpokládanou etiologií hydrocefalu u achondroplazie je zvýšený intrakraniální žilní tlak v důsledku stenózy jugulárního otvoru. Standardní léčbou je proto ventrikuloperitoneální shunting. U některých jedinců však může být prospěšná endoskopická třetí ventrikulostomie[ 17 ], což naznačuje, že mohou být zapojeny i jiné mechanismy, jako je obstrukce vývodu čtvrté komory v důsledku kraniocervikální stenózy.[ 18 ]
Stenóza kraniocervikálního spojení. Nejlepší prediktory potřeby subokcipitální dekomprese:
- Hyperreflexie nebo klonus dolních končetin
- Centrální hypopnoe na polysomnografii
- Zmenšení velikosti foramen magnum stanovené počítačovou tomografií kraniocervikálního spojení a porovnáno s normami u dětí s achondroplazií.[ 19 ]
- Jako další faktor, který je třeba zvážit při rozhodování o operaci, byly v poslední době navrženy důkazy o kompresi míchy a/nebo abnormality signálu váženého na T2.
Pokud se objeví jasné známky symptomatické komprese, je třeba pacienta urgentně odeslat k dětskému neurochirurgovi k provedení dekompresní operace. [ 20 ]
Léčba obstrukční spánkové apnoe může zahrnovat:
- Adenotonzilektomie
- Pozitivní tlak v dýchacích cestách
- Tracheostomie v extrémních případech
- Úbytek hmotnosti
Tyto intervence mohou vést ke zlepšení poruch spánku a určitému zlepšení neurologických funkcí.[ 21 ]
Ve vzácných případech, kdy je obstrukce natolik závažná, že vyžaduje tracheostomii, se k úlevě od obstrukce horních cest dýchacích používá operace posunu do střední části obličeje.[ 22 ]
Dysfunkce středního ucha. Časté infekce středního ucha, přetrvávající tekutina ve středním uchu a následná ztráta sluchu by měly být v případě potřeby léčeny agresivně. Doporučuje se dlouhodobé používání sond, protože jsou často potřebné do sedmi nebo osmi let věku dítěte.[ 23 ]
Pokud se problémy objeví v jakémkoli věku, doporučuje se použít vhodné léčebné metody.
Nízký vzrůst. Několik studií hodnotilo terapii růstovým hormonem (GH) jako možnou léčbu achondroplazie nízkého vzrůstu.[ 24 ]
Celkově tyto a další série vykazují počáteční zrychlení růstu, ale efekt se časem snižuje.
V průměru můžete očekávat nárůst výšky v dospělosti pouze o asi 3 cm.
Pro některé osoby zůstává možnost prodloužení končetin pomocí různých technik. Lze dosáhnout nárůstu výšky až o 30–35 cm. [ 25 ] Komplikace jsou časté a mohou být závažné.
Zatímco někteří doporučují provádět tyto zákroky již v šesti až osmi letech, mnoho pediatrů, klinických genetiků a etiků doporučuje odložit takovou operaci do doby, než se mladý člověk bude moci podílet na informovaném rozhodování.
Alespoň v Severní Americe se jen malá část postižených jedinců rozhodne podstoupit pokročilé prodlužování končetin. Lékařská poradní rada Little People of America vydala prohlášení týkající se použití pokročilého prodlužování končetin.
Obezita: Opatření k prevenci obezity by měla začít v raném dětství. Standardní léčba obezity by měla být u lidí s achondroplazií účinná, i když kalorická potřeba je nižší. [ 26 ]
Pro sledování pokroku by se měly používat standardní grafy hmotnosti a poměru hmotnosti k výšce specifické pro achondroplazii. Je důležité si uvědomit, že tyto křivky nejsou dokonalými křivkami poměru hmotnosti k výšce; byly odvozeny z tisíců datových bodů od lidí s achondroplazií.
Standardy indexu tělesné hmotnosti (BMI) byly vyvinuty pro děti ve věku 16 let a mladší. [ 27 ] BMI není standardizován pro dospělé s achondroplazií; srovnání s křivkami BMI pro průměrnou výšku povede k zavádějícím výsledkům. [ 28 ]
Varusová deformita. Doporučuje se každoroční ortopedické sledování buď odborníkem obeznámeným s achondroplasií, nebo ortopedickým chirurgem. Kritéria pro chirurgický zákrok byla publikována.[ 29 ]
Přítomnost progresivní symptomatické křivky vyžaduje odeslání k ortopedovi. Asymptomatická varusní deformita sama o sobě obvykle nevyžaduje chirurgickou korekci. Lze zvolit různé intervence (např. řízený růst s použitím osmi dlah, valgózní osteotomie a derotační osteotomie). Neexistují žádné kontrolované studie porovnávající výsledky léčebných možností.
Kyfóza. Kojenci s achondroplazií si často vyvinou flexibilní kyfózu. Existuje protokol, který pomáhá předcházet vzniku fixní úhlové kyfózy, a to včetně vyhýbání se flexibilním kočárkům, houpačkám a nosičům pro miminka. Doporučení proti sezení bez opory: při držení dítěte vždy vyvíjejte protitlak na záda.
- Kyfóza se u většiny dětí po zaujetí ortográdního držení těla a zahájení chůze významně zlepší nebo vymizí. [ 30 ]
- U dětí, u kterých nedojde k spontánní ústupu po zvýšení síly trupu a zahájení chůze, je obvykle dostatečná ortéza k zabránění přetrvávání torakolumbální kyfózy.[ 31 ]
- Pokud těžká kyfóza přetrvává, může být nutná operace páteře, aby se zabránilo neurologickým komplikacím.[ 32 ]
Spinální stenóza: Pokud se objeví závažné známky a/nebo příznaky spinální stenózy, je nutné neodkladné odeslání pacienta k chirurgickému specialistovi.
Obvykle se doporučuje rozšířená a široká laminektomie. Relevance zákroku závisí na úrovni (např. hrudní nebo bederní) a stupni stenózy. Pacienti měli lepší výsledky a zlepšenou funkci, čím dříve podstoupili operaci po nástupu příznaků [ 33 ].
Očkování: Nic ohledně achondroplazie nevylučuje všechna rutinní očkování. Vzhledem ke zvýšenému riziku respiračních onemocnění jsou obzvláště důležité vakcíny proti digestivnímu meningitidu, pneumokokové infekce a chřipce.
Adaptační potřeby: Vzhledem k nízké postavě mohou být nutné úpravy prostředí. Ve škole to může zahrnovat stoličky, snížené vypínače, toalety vhodné výšky nebo jiné prostředky přístupnosti, nižší lavice a opěrky nohou před židlemi. Všechny děti by měly být schopny v případě nouze samostatně opustit budovu. Malé ruce a slabé šlachy mohou ztěžovat jemnou motoriku. Mezi vhodné úpravy patří používání menší klávesnice, závaží pro pera a hladší povrchy pro psaní. Většina dětí by měla mít individuální vzdělávací program (IVP) nebo plán 504.
Pro jízdu jsou téměř vždy potřeba prodloužení pedálů. Mohou být také nutné úpravy pracovního místa, jako jsou nižší stoly, menší klávesnice, schůdky a přístup k toaletě.
Socializace: Vzhledem k velmi nápadnému nízkému vzrůstu spojenému s achondroplazií mohou mít postižení jedinci a jejich rodiny potíže se socializací a adaptací na školu.
Podpůrné skupiny, jako je Little People of America, Inc (LPA), mohou rodinám pomoci řešit tyto problémy prostřednictvím vzájemné podpory, osobního příkladu a programů sociálního povědomí.
Informace o zaměstnání, vzdělávání, právech osob se zdravotním postižením, adopci malých dětí, zdravotních problémech, vhodném oblečení, adaptivních pomůckách a rodičovství jsou k dispozici prostřednictvím celostátního zpravodaje, seminářů a workshopů.
Neexistuje žádný lék ani nefarmakologická léčba, která by dokázala tuto vrozenou vadu vyléčit.
Nejčastěji se používá fyzioterapie; léčba může být také nutná při hydrocefalu (pomocí shuntu nebo endoskopické ventrikulostomie), obezitě, [ 34 ] apnoe, [ 35 ] infekci středního ucha nebo spinální stenóze.
V některých klinikách se po dosažení věku dítěte pěti až sedmi let provádí chirurgická léčba: prodlužování kostí holenních, stehenních a dokonce i ramenních kostí nebo korekce deformity - za pomoci operací a speciálních ortopedických pomůcek - ve třech až čtyřech fázích, z nichž každá trvá až 6-12 měsíců.
Terapie je předmětem výzkumu
Podávání analogu natriuretického peptidu typu C prochází klinickými studiemi. První výsledky ukázaly, že je dobře snášen a u dětí s achondroplazií vede ke zvýšení rychlosti růstu oproti výchozímu stavu ( místo klinického hodnocení ). [ 36 ] Konjugovaný natriuretický peptid typu C je v současné době také prochází klinickými studiemi. [ 37 ] Dalšími faktory, které je třeba zvážit, jsou inhibice tyrosinkinázy [ 38 ], meklizin [ 39 ] a rozpustná rekombinantní lidská návnada FGFR3. [ 40 ]
Informace o klinických studiích pro širokou škálu onemocnění a stavů naleznete na webu clinicaltrials.gov v USA a v registru klinických studií EU v Evropě.
Prevence
Jediným preventivním opatřením je prenatální diagnostika vrozených onemocnění. [ 41 ], [ 42 ]
Předpověď
Jak dlouho se dožívají lidé s achondroplazií? Asi o 10 let kratší doba života, než je průměrná délka života.
Vzhledem k tomu, že patologické změny kostní tkáně a kloubů vedou k omezení sebeobsluhy a mobility, jsou dětem s touto diagnózou přiznáván status osoby se zdravotním postižením. Z dlouhodobého hlediska má většina pacientů normální prognózu, ale s věkem se zvyšuje riziko srdečních onemocnění. [ 43 ]