
Amyloidóza jater
Naposledy posuzováno: 07.06.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Amyloidóza je obvykle systémová, obecná patologie charakterizovaná akumulací amyloidu (specifický glykoprotein) v tkáních a následné narušení normální funkce orgánů. Amyloidóza jater je mnohem méně běžná než ledvina a slezina [1], ale téměř vždy doprovází systémové poškození těla. Žádná ze stávajících zobrazovacích technik nemůže konkrétně prokázat přítomnost amyloidu. I když je klinicky a radiologicky podezřelá, diagnóza amyloidózy závisí na biopsii tkáně, aby se potvrdila přítomnost amyloidních depozit. [3] Léčba je složitá, komplexní a zahrnuje imunosupresivní a symptomatická opatření. V závažných případech může být vyžadována transplantace jater.
Epidemiologie
Úspěch léčby přímo závisí na včasné diagnóze onemocnění, která způsobuje tvorbu komplexu protein-polysacharidů (amyloid) v různých orgánech a játrech. Jak ukazuje praxe, amyloidóza je obtížné předpokládat nebo podezřelé, i když je možné ji identifikovat a potvrdit. Skutečnost je, že ve více než 80% neuznaných případů je nemoc klinicky maskována jaterní patologií. Nejúčinnější diagnostickou metodou je biopsie.
Amyloidóza jater je vzácnějším problémem ve srovnání s amyloidózou ledvin. Současně jsou všechny případy jaterních lézí doprovázeny lézemi jiných orgánů. Patologie nejčastěji ovlivňuje převážně strukturální části jaterní trojice, která určuje minimální a nespecificita symptomatologie. Klinický a morfologický obraz hepatocelulárního nedostatku a portální hypertenze se projevuje v difúzní a intralobulární typu patologie.
Biopsie jater je oprávněná, když je přítomna hepatomegalie bez předchozích příznaků jater a v nepřítomnosti nefrotického syndromu.
Difúzní postižení jater je pozorováno asi 25% případů a u 75% pacientů jsou ovlivněny pouze portální trakty.
Primární amyloidóza ovlivňuje játra v 90% případů, zatímco sekundární amyloidóza ovlivňuje játra pouze v 47% případů.
Izolované postižení jater je velmi vzácné. Ledviny (asi 93%případů), slezina (72%), srdce (57%), pankreas (36%), nadledvinky (29%), střeva a plíce (každý 21%) jsou obvykle ovlivněny synchronně.
Ženy dostávají nemoc téměř dvakrát častěji jako muži. Průměrná délka života pacientů s amyloidózou je 52–64 let.
Příčiny jaterní amyloidóza
Amyloidóza postupuje s tvorbou a akumulací komplexního komplexu polysacharidového proteinu - amyloidu - v játrové tkáni. Problém výskytu k dnešnímu dni je nedostatečně studován. Pokud jde o sekundární patologii, její vzhled je obvykle spojen s takovými nemocemi:
- Chronické infekční procesy (tuberkulóza, syfilis, aktinomykóza);
- Purulentní zánětlivé procesy (mikrobiální endokarditida, osteomyelitida, bronchiektatické onemocnění atd.);
- Maligní onemocnění (leukémie, viscerální rakovina, lymfogranulomatóza).
Reaktivní forma amyloidózy se nachází u pacientů se souběžnou aterosklerózou, revmatologickými chorobami (Bechterewova choroba, revmatoidní artritida), psoriázou, chronickými zánětlivými a multisystémovými procesy (včetně sarkoidózy). Hlavní rizikové faktory: dědičná predispozice, poruchy buněčné imunity, hyperglobulinémie.
Patogeneze
Existuje řada předpokladů týkajících se původu amyloidózy jater. Většina specialistů dodržuje verzi dysproteinózy, imunologické a mutační povahy nemoci a také místní buněčné geneze. Verze buněčné geneze zahrnuje změny v reakcích pracujících na buněčné úrovni (tvorba fibrilárních prekurzorů amyloidu komplexem makrofágů), ačkoli je amyloid tvořen a hromadí se mimo buněčné struktury.
Verze dysproteinózy je založena na skutečnosti, že amyloid je produktem nesprávného metabolismu proteinu. Základní patogenetická vazba problému spočívá v dysproteinemii a hyperfibrinogenemii, která vede k akumulaci hrubého rozptýleného proteinu a paraproteinových frakcí v plazmě.
Podle imunologické verze je tvorba amyloidů způsobena reakcí antigenu a protilátky, kde produkty rozpadu tkáně nebo cizí proteiny působí jako antigeny. Amylulace amyloidů se nachází hlavně v oblasti tvorby protilátek a nadměrné přítomnosti antigenů.
Vědci nejpravděpodobnější verze vědci zvažují teorii mutace, která zohledňuje řadu mutagenních faktorů, které mohou vést k abnormalitám při syntéze proteinů.
Amyloid je komplexní hypoprotein, který se skládá z globulárních a fibrilárních proteinů kombinovaných s polysacharidy. Amyloidní akumulace ovlivňují intimu a adventii vaskulární sítě, stroma parenchymatózních orgánů, struktura žláz atd. Amyloidní amymulace nezpůsobují funkční poškození. Malé akumulace nezpůsobují funkční poruchy, ale s intenzivní amyloidní přítomností zvýšení jater v objemu, mění vzhled orgánu, vyvíjí nedostatek funkce.
Amyloidóza jater je charakterizována depozicí amyloidních fibril v prostoru dysse, která obvykle začíná v periportální oblasti, ačkoli je někdy centrilobulární a může také ukládat jaterní vaskulaturu. [4], [5] V závažných případech vede depozice amyloidů k tlakové atrofii hepatocytů, která zabraňuje průchodu žluči, což má za následek cholestázi nebo může blokovat sinusoidy, což má za následek hypertenzi portálu. [6], [7], [8]
Symptomy jaterní amyloidóza
Klinický obraz v amyloidóze jater je rozmanitý, záleží na intenzitě akumulace amyloidů na jeho biochemických rysech, trvání patologického procesu, stupně poškození orgánů a porušování jejich funkčního stavu.
V latentním stádiu amyloidózy, kdy lze amyloidní amymulace v játrech detekovat pouze mikroskopickým vyšetřením, chybí první známky onemocnění. S dalším vývojem a zvyšováním funkčního deficitu orgánu postupuje symptomatologie.
Játra postupně zhoustne a zvětšuje. Metoda palpace může být hmataná změna, ale hladké a bezbolestné hranice orgánu. Zřídka je patologie doprovázena bolestí v subkostální oblasti na pravé straně, dyspepsií, zvětšením sleziny, žloutnutí kůže, sliznicemi a skleáry, hemoragickým syndromem.
Nejuznávanější příznaky u amyloidózy jater: [9], [10]
- Amyloidní akumulace v játrech způsobuje hepatomegalii u 33-92% pacientů;
- Mírná žloutenka
- Hypertenze portálu;
- Mírná až těžká cholestáza.
Protože amyloidóza velmi zřídka ovlivňuje pouze jeden orgán, je obvykle přítomna další symptomatologie:
- Když poškození ledvin vyvine nefrotický syndrom a arteriální hypertenzi s dalším selháním ledvin, otoků, někdy trombózy ledvin, leukocyturie, hematurie, hypoproteinémie, azotemie atd.
- Když je ovlivněno srdce, vyvíjí se stav podobný restriktivní kardiomyopatii (poruchy rytmu, kardiomegalie, zvyšování srdečního deficitu, slabost a dušnost, otoky, méně často - akumulace tekutin v břišní a pleurální dutině, perikarditida);
- Pokud je ovlivněn trávicí trakt, může dojít k makroglossii, slabosti a peristaltize jícnu, nevolnost a pálení žáhy, zácpa nebo průjem atd.;
- Když je ovlivněna pankreas, jsou přítomny příznaky chronické pankreatitidy;
- Pokud se jedná o muskuloskeletální mechanismus, vyvíjejí se symetrická polyartritida, syndrom karpálního tunelu, myopatie, a pokud je ovlivněn nervový systém, nacházejí se polyneuropatie, ochrnutí, ortostatický nízký krevní tlak, zvýšené pocení, demenci.
Pokud se patologická reakce šíří na kůži, objeví se na obličeji, krku, krku, krku, krku, krku. Je možný obrázek neurodermatitidy, červené spinocelusíční horečky, sklerodermie.
Kombinace více amyloidních lézí a rozmanitosti symptomatologie ztěžuje identifikace amyloidózy jater a vyžaduje komplexní a úplnou diagnózu.
Formuláře
Podle klasifikace WHO se rozlišuje pět typů amyloidózy:
- Al (primární);
- AA (sekundární);
- Attr (dědičný a senilní systémový);
- Ap2M (u pacientů na hemodialýze);
- AIAPP (u pacientů s diabetes mellitus nezávislý na inzulínu);
- AB (pro Alzheimerovu chorobu);
- AANF (senilní síň amyloidóza).
Existuje lokální amyloidóza jater, ale častěji se jedná o systémovou lézi, ve které patologický proces zahrnuje také ledviny, srdce, slezinu, nervový systém, jakož i další orgány a tkáně.
Komplikace a důsledky
Systémová amyloidóza postupně vede k vývoji akutních patologických procesů, které mohou zase vést k smrti. Mezi nejčastější a život ohrožující komplikace patří:
- Časté infekční (bakteriální, virové) patologie, včetně pneumonií, pyelonefritidy, glomerulonefritidy;
- Chronické selhání jater a ledvin;
- Chronické srdeční selhání (může předcházet infarktu myokardu);
- Hemoragické tahy.
Žilní trombóza se vyskytuje v důsledku akumulace a ukládání proteinů na žilních stěnách. Vyvíjí se onemocnění postižených cév, které se zužují, vyvíjí se selhání orgánů. V průběhu času, na pozadí dlouhodobé hyperproteinémie, může plavidlo zcela uzavřít. Jakákoli z komplikací může vést k nepříznivému výsledku - smrti.
Diagnostika jaterní amyloidóza
Pokud je podezření na amyloidózu jater, diagnostická opatření se provádějí po povinných konzultacích, gastroenterologem i terapeutem a revmatologem, kardiologem, dermatologem, neurologem, urologem. Je důležité komplexně vyhodnotit údaje o anamneze a klinických projevech, provádět komplexní laboratorní a instrumentální diagnostiku.
Testy nutně zahrnují vyšetření moči a krve. V jaterní amyloidóze je často nalezena kombinace leukocyturie s proteinurií a cylindrurií a hypoproteinémie - s hyperlipidémií, anémií, hyponatrémií a hypokalcemií, snížena počet destiček. Paraproteiny jsou detekovány elektroforézou v moči a v séru.
Instrumentální diagnostika zahrnuje:
- EKG, Echo;
- Břišní ultrazvuk;
- Rentgenové paprsky žaludku, jícnu;
- Irigografie, rentgenové paprsky barya;
- Endoskopie.
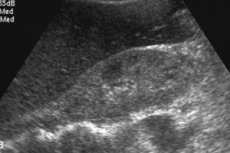
Radiologické nálezy jaterní amyloidózy zahrnují nespecifickou hepatomegalii, zvýšená echogenita na ultrazvuku nebo hustotě na počítačové tomografii (CT) a zvýšená intenzita signálu T1 při zobrazování magnetickou rezonancí (MRI). [12] Scintigrafie s indikátory souvisejícími s TC-99M ukazuje heterogenní vychytávání, ale je nespecifická. [13], [14] Ukázalo se, že GC zvyšuje tuhost jater měřenou elastografií; [15], [16], [17], ale existuje jen několik kazuistik. Magnetická rezonance elastografie (MRE) je v současné době nejpřesnější neinvazivní metodou pro detekci a fázi jaterní fibrózy, [18], [19] MRE je užitečná pro detekci progrese, reakce na léčbu a predikci dekompenzace jater u pacientů s fibrózou jater. [20]
Je obtížné určit amyloidózu jater na ultrazvuku: je stanoveno zvětšení orgánu, přičemž nejvíce specifičtější hepatomegálie přesahující 15 cm. Pod kontrolou ultrazvuku se provádí biopsie, která se stává určujícím indikátorem diagnózy. Pomocí speciální jehly se odebírá malé množství jaterní tkáně, poté je obarveno speciálním barvivem a zkoumáno pod mikroskopem, což vám umožňuje přímo vidět amyloidní depozity.
Definitivní diagnóza se provádí až po detekci amyloidních fibril v tkáni jater a dalších orgánů. Geneticky stanovený typ amyloidózy je stanoven pečlivým geneticky léčeným vyšetřením rodokmenu.
Diferenciální diagnostika
Amyloidóza by měla být podezřelá u všech pacientů s kombinací renální proteinurie, restriktivní kardiomyopatie, autonomní nebo periferní neuropatie a hepatomyelií, a to i v nepřítomnosti monoklonálního paraproteinu. Ověření typu amyloidózy je velmi důležité, protože léčba lézí různých etiologií je velmi odlišná.
Histologická diagnóza zahrnuje zbarvení kongo červenou následovanou mikroskopickým vyšetřením v polarizačním světle. Je vhodné biopsie několik vzorků tkání najednou. Pokud se výsledek zbarvení stane pozitivním, provádí se imunohistochemická analýza s použitím monoklonálních protilátek k prekurzorovým proteinům k identifikaci typu amyloidu.
Analýza DNA se provádí pro rozlišení mezi primární amyloidózou a různými variacemi geneticky stanovené amyloidózy. Amyloidní fibrily mohou být izolovány ze vzorků biopsie a sekvestrovány do jednotlivých aminokyselin.
Další studie pro stanovení dyscrazie plazmatických buněk:
- Elektroforéza sérových proteinů krve a moči;
- Imunotest pro volné světelné řetězce;
- Imunofixace (imunoblotting) sérových proteinů;
- Aspirace kostní dřeně a trepanobiopsie.
Diagnóza amyloidózy jater je zdlouhavý a pracovní proces, který vyžaduje zvýšenou pozornost odborníků a kvalitní vybavení klinik a laboratoří.
Kdo kontaktovat?
Léčba jaterní amyloidóza
Cílem léčebných opatření je snížit koncentraci již existujících amyloidních proteinů v krvi (eliminující příčinu amyloidózy) a na podporu přiměřené funkce jater.
Sekundární amyloidóza vyžaduje blokování zánětlivého procesu (u chronických infekčních a autoimunitních patologií). U autoimunitních onemocnění se doporučuje používání cytostatiky. Pro odstranění chronických infekčních procesů je oblast zánětu často chirurgicky odstraněna. Tento přístup může často zastavit další progresi amyloidózy a zlepšit funkci jater.
Primární amyloidóza vyžaduje použití chemopreventivních léčiv a někdy transplantace kostní dřeně.
Současné pokyny doporučují kombinaci cyklofosfamidu, bortezomibu, dexamethasonu (Cybord) a daratumumabu jako terapie první linie u pacientů nově diagnostikovaných s AL.
Bortezomib je inhibitor proteazomu. Proteazomy se podílejí na snižování proteotoxicity a regulaci proteinů, které řídí buněčnou progresi a apoptózu. Plazmatické buňky, které vytvářejí amyloid, jsou zvláště citlivé na inhibici proteazomu, protože se spoléhají na proteazom, aby se snížily toxické účinky světelných řetězců a zabránily apoptóze.
Daratumumumab je monoklonální protilátka (MAB), která se váže na CD38, transmembránový glykoprotein exprimovaný na povrchu plazmatických buněk, což indukuje apoptózu. Je to jediný lék speciálně schválený pro léčbu al amyloidózy při použití u Cybordu. Účinnost Cybord-Daratumumumabu je velmi vysoká, přičemž 78% pacientů dosáhne významné hematologické odpovědi (definované jako úplná odpověď nebo velmi dobrá částečná odpověď). Střední přežití u malé skupiny pacientů, kteří dostávali Cybord (n = 15), bylo 655 dní ve srovnání s 178 dny u pacientů, kteří dostávali jinou léčbu na bázi melfalan-dexamethason (n = 10). 4
Tyto terapie však mají četné vedlejší účinky, včetně kardiotoxicity, což vede k potřebě snižování dávky nebo suspenzi léčby a použití dalších méně účinných, ale tolejších terapeutických strategií.
Isatuximab, monoklonální protilátka proti CD38 podobné daratumumabu, se studuje pro léčbu dyscrazie plazmatických buněk, která je základem AL.
V současné době se studují tři monoklonální protilátky Birtamimab, CAEL-101 a AT-03 pro odstranění amyloidních fibril z nemocných orgánů. Výsledky těchto studií budou schopny nabídnout přímé důkazy pro hypotézu, že odstraněním fibril depozice lehkého řetězce z orgánů dochází ke zlepšení funkce orgánů. [21]
Za účelem podpory jaterní funkce jsou předepisovány léky založené na kyselině urso-deoxycholové (příklad - ursosan). Urso-deoxycholová kyselina pomáhá stabilizovat buněčné membrány, snižuje nepříznivý účinek toxických mastných kyselin v žlučové stázi vyvolané depozity amyloidů a pomáhá obnovit normální odtok žluči.
Kromě toho symptomatická terapie a podpora fungování jiných vitálních struktur, jako je nervový systém, srdce, ledviny atd. Podpůrná terapie u pacientů s jaterní amyloidózou zahrnuje různé klinické aspekty, včetně léčby srdečního selhání, arytmie, poruchy vedení, vedení vodivosti, poruchy vedení, vodivosti, poruchy vedení, vedení vodivosti, poruchy vedení, vedení vodivosti.
Jiná ošetření závisí na typu amyloidózy a na které části těla jsou ovlivněny. Ošetření může zahrnovat: [22]
- Léky, které zmírňují příznaky, jako jsou léčiva bolesti, léky na nevolnost nebo léky, které snižují otoky (diuretika);
- Léky ke snížení amyloidu;
- Dialýza ledvin;
- Transplantace jater.
Játra produkují 95% TTR (transthyretin, protein zapojeného do transportu tyroxinu (T4) a protein vázající retinol. Transtryretin je syntetizován hlavně v játrech a je bohatý na beta řetězce, které mají tendenci agregovat do inkoluálních amyloidních fibril) měřených v seru. Proto byla transplantace jater historicky (od roku 1990) navržena jako terapie první linie k eliminaci hlavního zdroje amyloidogenního TTR u pacientů s familiární formou (ATTRV), zatímco není indikována ve formě ATTR-WT. Transplantace jater mladých pacientů v raných stádiích onemocnění je spojena s vysokou 20letou mírou přežití. Transplantace jater se zdá být účinnější v některých mutacích a u jiných méně účinná, jako je V122i (spojená s kardiomyopatií). Kombinovaná transplantace jater a srdce je také možná u mladých pacientů s kardiomyopatií a údaje o literatuře o malé skupině pacientů naznačují, že tato kombinace má lepší prognózu než transplantace srdce.
Pacienti s jaterní amyloidózou jsou kontraindikovaní užívající srdeční glykosidy a antagonisty vápníku, jako je diltiazem nebo verapamil, které se mohou akumulovat v amyloidu. Inhibitory ACE a beta-adrenoblockers se používají s opatrností.
V ortostatické hypotenzi jsou předepsány mineralokortikoidy nebo glukokortikosteroidy, s přihlédnutím k dekompenzaci srdečního selhání. Alfa-adrenomimetický midodrin (gutron) se také používá s opatrností.
V neuropatiích jsou vhodné antikonvulzivy a antidepresiva.
V některých případech amyloidózy jater musí lékaři zvážit transplantaci orgánu.
Prevence
Vzhledem k nedostatku informací o patogenezi amyloidózy jater nemohou specialisté vyvinout specifickou prevenci nemoci. Hlavní úsilí je proto sníženo na včasnou detekci a léčbu jakýchkoli chronických patologií, které mohou vyvolat rozvoj poruchy. Pokud existují případy amyloidózy jakékoli lokalizace v rodině, doporučuje se systematicky navštěvovat lékaře pro výdejní zkoušky.
Obecně jsou preventivní opatření snížena na včasné odstranění infekčních chorob, zejména těch, které mají tendenci se transformovat na chronický proces. Jde o prevenci vývoje tuberkulózy, plicních infekcí atd. Je důležité včas detekci a přiměřenou léčbu streptokokových infekcí, které se mohou stát příčinou chronických forem autoimunitních zánětlivých procesů. Mluvíme o Scarlatině, streptokokové tonzilitidě atd.
Pokud pacient již má autoimunitní onemocnění, měl by systematicky konzultovat s lékařem, pozorovat aktivitu patologie, aplikovat nezbytné léky, jak je předepsáno lékařem, upravte dávky podle indikací.
Předpověď
Prognóza pacientů s jaterní amyloidózou je nepříznivá. Onemocnění se zvyšuje pomalu, ale nepřetržitě, což nakonec způsobuje dysfunkci postižených orgánů a letálního výsledku - zejména kvůli selhání orgánů.
Pacienti se systémovou patologií umírají hlavně v důsledku vývoje chronického selhání ledvin, i když v některých případech hemodialýza nebo kontinuální ambulantní peritoneální dialýza zlepšuje prognózu těchto pacientů. Míra přežití pacientů na hemodialýze, bez ohledu na její typ, lze porovnat s mírou lidí s jinými systémovými patologiemi a diabetes mellitus.
Hlavní příčinou úmrtí během hemodialýzy je vývoj komplikací z kardiovaskulárního systému.
Transplantace jater byla dlouho považována za jednu z hlavních metod léčby onemocnění a nejoptimističtější míra přežití je pozorována u pacientů, jejichž věk nepřesahuje 50 let (za předpokladu, že patologický proces je krátkodobý a index tělesné hmotnosti je normální). Pacienti s amyloidózou jater v kombinaci s periferní neuropatií mají poněkud horší prognózu.