
Hamartoma
Naposledy posuzováno: 07.06.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
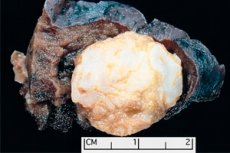
Tvorba podobná nádoru lokalizovaná v jakékoli anatomické oblasti vyplývající z abnormálního růstu benigní tkáně, v medicíně je v medicíně definována jako hamartoma (z řecké Hamartie - chyba, defekt). [1]
Epidemiologie
Statisticky hamartomy představují 1,2% benigních novotvarů. Odhaduje se, že prevalence plicních hamartomů je přibližně 0,25% obecné populace a představuje až 8% všech plicních novotvarů. Většina plicních hamartomů je diagnostikována mimochodem u pacientů ve věku 40 až 70 let, ale je velmi vzácná v pediatrické praxi.
Obecně platí, že většina hamartomů je diagnostikována u mužů, ačkoli v ledvinách jsou běžnější u žen a jsou identifikovány ve středním věku.
Asi 5% benigních nádorů prsu jsou hamartomy a nejčastěji ovlivňují ženy starší 35 let.
80-90% hamartomatózních lézí mozku a více než 50% hamartomů srdce je spojeno s hlízočnou sklerózou.
Příčiny hamartomy
Gamartomas patří k vrozeným malformacím a jsou formace benigního charakteru, které jsou tvořeny z mezenchymálních tkání pocházejících z zárodečných listů. A příčiny jejich výskytu jsou spojeny s nekontrolovaným buněčným dělením cytologicky normálních tkání (pojivové, hladké svalstva, tuk nebo chrupavka), charakteristickým pro dané anatomické umístění a jejich fokálním přerůstání během embryogeneze téměř jakéhokoli orgánu nebo anatomické struktury.
Výskyt více hamartomů u stejného pacienta je často označován jako hamarmatóza nebo pleiotropický hamartoma.
Tyto nádory se mohou vyskytnout sporadicky nebo v přítomnosti určitých autozomálně dominantních zděděných onemocnění a geneticky určených syndromů.
V mnoha případech se hamartomy tvoří, když se vzácné genetické onemocnění multisystémové povahy - tuberózní sklerózy -projevuje krátce po narození nebo při reklinghausenově familiární chorobě - neurofibromatóza typu 1. [2]
Rizikové faktory
Mezi hlavní rizikové faktory pro tvorbu hamartomu patří přítomnost tzv. Genetických syndromů hamartomatózní polypózy v historii pacientů, včetně:
- Syndrom mnohočetného hamartomu- Cowdenův syndrom, ve kterém se vytváří více hamartomů ecto-, ento- a mezodermálního původu, jsou pozorovány gastrointestinální polypózu a mukokutánní projevy;
- Syndrom Peutz-Jeghers-turen (charakterizován vývojem benigních hamartomatózních polypů v gastrointestinálním traktu);
- Proteus Syndrom;
- Weil syndrom - juvenilní polypóza tlustého střeva;
- Syndrom Bannayan-Riley-Ruvalcaba, který stejně jako syndrom Cowdena produkuje několik hamartomů (hamartomatózní polypy) střeva;
- Carney-Stratakisův syndrom a Carney Complex.
Kromě toho se hamartomy tvoří u pacientů s dědičným Watsonovým syndromem a v případech sporadicky se vyskytujícího nebo vrozeného syndromu Pallister-Hall s hypotalamickým hamartomem a polydactyly.
Patogeneze
Mechanismus zvýšené proliferace zárodečných tkání s tvorbou malformací podobných nádorům v různých orgánech je vysvětlen chromozomálními aberacemi a genovými mutacemi, které se mohou vyskytnout spontánně nebo zdědit.
U tuberózní sklerózy byly identifikovány mutace v genech TSC1 nebo TSC2 - nádorové supresory, které brání a inhibují nadměrnou proliferaci - příliš rychlou nebo nekontrolovanou buněčnou růst a dělení -. A u neurofibromatózy typu 1 a Watsonův syndrom - zárodečný mutace mitochondriálního nádorového supresorového genu NF1.
U nádorového syndromu hamartomu, který kombinuje Cowden, Protea, Bannayan-Riley-Ruvalcaba a syndromy juvenilní polypózy, je patogeneze spojena s mutací genu PTEN, který kóduje enzym zapojený do regulace proliferace a je považován za nádorový potlačovací gen.
Mutace v genu STK11 kódující strukturu a funkci jednoho z transmembránových serinových enzymů, které snižují jeho schopnost omezit dělení buněk, vedou k peutz-jegherův-turenovým syndromu s vývojem střevních polypů a pigmentovaných kožních lézí. U syndromu Pallister-Hall byla identifikována mutace v genu GLI3, transkripční faktor zapojený do tvorby intrauterinní tkáně.
Nekontrolovaný růst buněk v důsledku mutací genu tedy vede k tvorbě hamartomu.
Symptomy hamartomy
V závislosti na lokalizaci hamartomů se jejich typy rozlišují a každá z nich má svou vlastní strukturu a symptomatologii.
Hamartoma plic
Plicní hamartoma se může tvořit v jakémkoli laloku a periferních částech plic a skládá se z normálních tkání přítomných v plicích: tukové, epiteliální, vláknité a chrupavkové. V 80% případů převládá složka chondroidní (hyalinní chrupavkové buňky) zahrnutím adipocytů - tukové tkáňové buňky a epiteliálními buňkami dýchacích cest. [3]
Dřívější jména: Chondroid Hamartoma, Mezenchomoma, chondromatózní hamartoma nebo Hamartochondroma se v současné době nedoporučují.
Na druhé straně je mezenchymální cystický hamartoma plic méně běžný a u většiny pacientů je spojen se syndromem Cowdena.
Hamartomatózní léze plic se nemusí projevit, ale může způsobit příznaky ve formě chronického kašle (často s hemoptysis), síť při dýchání a potíže s dýcháním. [4]
Hamartoma srdce
Benigní primární srdeční nádory u dospělých zahrnuje zralý hamartom myocytů a u kojenců a dětí s tuberózní sklerózou, rhabdomyomem, tj. Myokardiálním hamartomem komor nebo interventrikulárního septa. [5]
Zralý kardiomyocytový hamartoma se vyvíjí ve ventrikulární stěně (a zřídka v atria) a může se objevit jako více lézí, které jsou husté hmotnosti úzce spojené s podkladovým myokardem. Nádor může způsobit příznaky srdečního selhání: bolest na hrudi, palpitace a arytmie, srdeční šelest, otoky, dušnost, cyanóza.
Srdeční rhabdomyomy, z nichž většina je diagnostikována v prvním roce života, se skládají ze srdeční svalové tkáně tvořené embryonálními myoblasty a mají vzhled pevných ohniskových hmot bez kapsle.
Typicky tyto hamartomy představují asymptomaticky a spontánně ustupují před věkem 4 let.
Někteří odborníci také považují hamartomatózní léze, které jsou také spojeny s komplexem Carney myxoma srdce. [6]
Gamartoma gastrointestinálního traktu
Žaludeční hamartoma je mezenchymální hmota ve formě epiteliálního hyperplastického polypu žaludku, peutz-jeghers polyp a vzácný myoepiteliální hamartoma-s hypertrofickými hladkými svalovými svaly. Mezi další jména pro tento hamartoma patří myoglandulární hamartoma, adenomyomatózní hamartoma a žaludeční adenomyom. Mezi typické klinické projevy patří dyspepsie, epigastrická bolest a krvácení z horního GI. [7], [8]
Více informací v materiálu - žaludeční polypóza
Střevní hamartoma je hamartomatózní nebo hyperplastický polyp tlustého střeva, diagnostikován jako adenomatózní nebo tubulární adenom. Když je hamartoma lokalizován v brunnerově žláze duodenum, symptomy se projevují bolestí v epigastrické oblasti; nevolnost, zvracení a nadýmání (označující střevní obstrukci); a pokud mají značnou velikost, gastrointestinální krvácení. V případech myoepiteliálního hamartomu ileum si pacienti stěžují na bolest břicha, sníženou tělesnou hmotnost a vyvinou chronickou anémii. [9], [10]
Také si přečtěte - rektální polypy
Retrorektální hamartoma je cystický hamartoma nebo vícechovanou cystu retrorektálního prostoru (volná pojivová tkáň mezi konečníkem a vlastní fasádou), ke kterému se nejčastěji vyskytuje u žen středního věku. Má vzhled cysty vyboulené ze zadní stěny konečníku, který je lemován epitelem a obsahuje chaoticky uspořádaná vlákna hladkého svalstva. Tento hamartoma představuje nižší bolest břicha a opakující se zácpa. [11], [12]
Hamartomas jater a sleziny
Vícenásobná biliární hamartoma játra je hamartoma interdollického intrahepatického žlučovodů spojených s malformacemi jejich vývoje během embryonálního období. Tento hamartoma (jeden nebo více) sestává z náhodně rozšířených shluků žlučovodů a fibrokolagenní stromy. [13]
Biliární hamartomy jsou asymptomatické a obvykle se objevují náhodně (během radiologického vyšetření nebo laparotomie). [14]
Vzácný a často náhodně detekovaný primární novotvar benigního charakteru je hamartoma sleziny, která se skládá z prvků červené buničiny sleziny - ve formě dobře definované homogenní hmoty pevné konzistence. Tato malformace může být jednorázová nebo vícenásobná; Při stisknutí splenického parenchymu může v levé subkostalní oblasti existovat pocit nepohodlí a bolesti. [15], [16]
Renální hamartomy
Nejběžnější hamartoma ledviny je diagnostikována jako angiomyolipom ledviny, protože tento benigní nádor sestává ze zralé tukové tkáně se zabudovanými vlákny hladkého svalstva a krevních cév. Tvoří se v hlízkové skleróze u 40-80% případů. Zvětšení velikosti hamartomu (více než 4-5 cm) vede k bolesti a vzhledu krve v moči. [17], [18]
Hamartoma prsu
Diagnostické definice hamartomu přijetí WHO jsou pojmy jako adenolipom, chondrolipom a myoidní hamartoma. Ačkoli se často nazývá fibroadenolipom mamology, protože tvorba nádoru obsahuje buňky vláknité, žlázové a tukové tkáně uzavřené v tenké pojivové tkáňové tobolce s odlišnými obrysy. Při vizualizaci lze pozorovat fokální kalcifikace. V tomto případě klinické projevy chybí. [19], [20]
Také si přečtěte - nádory prsu
Hamartomas mozku
Jedna třetina pacientů s hlízou sklerózou má mozkový hamartoma ve formě intrakraniálního kortikálního růstu nebo hlíz v různých lalocích - na hranici šedé a bílé hmoty - nebo subependymální uzly podél stěn mozkové komory. Může se také tvořit astrocytický hamartoma, subependymální obří buněčné astrocytom s kortikálním narušením, dysmorfní neurony a velké gliové buňky mozkového parenchymu (astrocyty). Mezi příznaky mozkových hamartomů patří útoky na záchvaty a mentální retardace u dětí. [21], [22]
Vzácná malformace, která se vyskytuje během embryogeneze a je přítomna při narození, je hypothalamický hamartoma, což je hmotnost heterotopických neuronů a gliových buněk. Jak roste mozek dítěte, nádor se zvětšuje, ale nerozšíří se do jiných mozkových oblastí. [23], [24]
Pokud se v přední části hypothalamusu (tuber cinereum) vytvoří hypertrofované tkáně, kde se k němu hypofyzá. Časná ochlupení na vlasy a hlasové mutace u chlapců.
Když se hamartomy tvoří v zadní části hypothalamu, mohou existovat abnormality v elektrické aktivitě mozku, která se v raném dětství projevuje záchvaty a v pozdějším stádiu (ve věku 4 až 7 let) epilepsií a záchytnými záhadami a agresivními a agresivními a agresivními látkami, a agresivními a agresivními látkami a agresivitami, a také agresivními a agresivními záhadami.
Hypofýza hamartoma je sporadicky vyskytující se benigní hypofýza.
Dospělí středního věku se syndromem Cowdena mohou mít vzácnou hmotu podobnou nádoru, hamartoma mozečku, diagnostikovaná jako dysplastická cerebelární gangliocytom nebo onemocnění lhermitte-duclos. Příznaky mohou chybět nebo se projevit jako bolest hlavy, závratě, zhoršená koordinace pohybů a ochrnutí jednotlivých lebečních nervů.
Hamartoma lymfatických uzlin
Když buňky hladkého svalstva a tukové tkáně, jakož i krevní cévy a kolagenní stroma inguinálních, retroperitoneálních, submandibulárních a cervikálních lymfatických uzlin, se vytvoří angiomyomatózní hamartom lymfatického uzlinu nebo se tvoří částečný nebo kompletní nahrazení jeho parinchym. [25], [26]
Hamartoma kůže
V přítomnosti hlízové sklerózy nebo neurofibromatózy jsou pozorovány různé hamartomy kůže, nejčastěji ve formě hypopigmentovaných skvrn; Skvrny kávy a mléka; angiofibroma (na tvářích, bradě, nasolabiální záhyby); Shagreen skvrny různých lokalizací (což jsou pojivové tkáně Nevi); vláknité plaky na čele, pokožku hlavy nebo krku.
Vzácným dermatologickým projevem hlízové sklerózy (zejména u mužů) je folikulocystický a kolagen hamartoma, charakterizovaný hojným ukládáním kolagenu v dermis, soustřednou perifolikulární fibrózu a keratinem naplněným podkutánními cysty na histopatologickém vyšetření. [27]
Na hamartomy sestávající z melanocytů (buňky, které produkují pigmentový melanin), většina odborníků také odkazuje na různé melanocytární novotvary, zejména vrozené melanocytické nevi, které představují abnormalitu embryogeneze.
Pokud jde o etiologii, hamartomy složené z vaskulární tkáně jsou také hemangiomy kůže.
Pacienti se syndromem Peutz-Jeghers-Thuren mají hamartoma ve formě nerovnoměrné pigmentace kůže a sliznic - lentiginóza periorificialis
Případy lineárního papulárního ektodermálního mezodermálního hamartomu (Hamartoma moniliformis) ukazují lineární papulární vyrážku z masa na hlavě, krku a horní hrudi.
A sebocytický hamartoma je hamartoma mazových žláz, přečtěte si více v publikaci - sebaceous nevus.
Hamartoma oka
Pigmentované hamartomatózní léze duhovky u neurofibromatózy typu 1 a Watsonova syndromu - ve formě nodulárních shluků dendritických melanocytů - jsou definovány jako hamartomy Iris nebo Lisch. Jsou průhledné (obvykle neovlivňují vidění) zaoblené žlutohnědé papuly ve tvaru kupole, které vyčnívají nad povrch IRIS.
A pacienti s juvenilním angiofibromem nosofarynxu a familiární adenomatózní polypóza často vyvíjejí kombinovaný hamartoma epitelu sítnice a pigmentu sítnice-ve formě černé skvrny na centrální (makulární) části retiny. [28]
Hamartoma nosu
Nosní hamartoma je definován specialisty jako nosní chondromesenchymální hamartoma nebo nosní chondroma, kvůli benignímu proliferaci respiračního epitelu, submukózních žláz a chondro-kostní mesenchym. Jeho klinické projevy závisí na velikosti a lokalizaci léze a zahrnují: nosní přetížení, potíže s nosním dýcháním a kojením u kojenců, čistého vodnatého výtoku nosní a nosní krvácení. Hamartoma může růst s dítětem a šířit se do očních oběžných drah, což má za následek přesun oční bulvy, strabismu nebo okulomotorické poruchy. [29]
Hamartoma u dítěte
Všechny výše uvedené hamartomatózní léze různých orgánů a anatomických struktur jsou přítomny u dětí s odpovídajícími syndromy.
Novorozenci jsou přítomni s mesenchymálním hamartomem hrudní stěny nebo chrupavkového hamartomu žebra, což jsou pevné imobilní hmoty vyplývající z ohniskového přerůstání normálních kosterních prvků s chrupavkou, vaskulárními a mezenchymálními prvky. Tento hamartoma může způsobit respirační selhání a rozvoj syndromu respirační tísně. Mezenchymální hamartoma játra je druhý nejčastější benign jaterní nádor u dětí. Tato tvorba podobná nádoru (častěji lokalizovaná v pravém laloku orgánu) sestává z buněk mezenchymální stromy, hepatocytů a epiteliálních buněk obložení žlučovodů. Klinický obraz zahrnuje hmatatelnou hmotu v břišní dutině, anorexii a úbytek hmotnosti a v případě významné velikosti (až 10 cm a více) nádor pokrývá extrahepatické žlučovody a dolní vena cava, což vede k žloutenku a edému dolních končetin.
Hamartoma je vrozený mesoblastický nefrom (vyskytující se u 1 u 200 000 kojenců), který může vést k břišnímu nadýmání novorozence s hmatatelnou hmotou husté konzistence v pravém horním kvadrantu břicha. Kojenci mohou také vystupovat rychlé mělké dýchání.
Mezi vzácné vrozené anomálie patří vláknitý hamartoma dětství, který se vyskytuje u dětí v prvních dvou letech života a představuje se jako bezbolestná nodulární hmota v subkutánních tkáních axily, krku, ramene a předloktí, zad a hrudníku, stehna, nohy a vnější genitálie.
Ekcrin angiomatózní hamartoma u dítěte může být přítomen při narození nebo se projevit v raném dětství. Tento benigní nádor hamartomatózní přírody má obvykle vzhled namodralých nebo nahnědlých uzlů a/nebo plaků, které jsou výsledkem proliferace tkáně ekcrinní potové žlázy a kapilár ve střední a hluboké vrstvě dermis. Tento hamartoma může způsobit lokalizovanou hyperhidrózu a zvýšený růst vlasů.
Komplikace a důsledky
Obecně se souhlasí s tím, že hamartomy se zřídka opakují nebo se transformují na maligní nádory. Často vykazují malé nebo žádné příznaky a někdy dokonce v průběhu času mizí. Ale ve závažnějších případech a v závislosti na místě tvorby mohou tyto malformace mít vážné komplikace a důsledky.
Za prvé, hamartoma může růst do takové velikosti, že začne tlačit na okolní tkáně a orgány a narušit jejich funkce.
Srdeční hamartoma u dětí může vést k přetrvávajícím abnormalitám srdečního rytmu, defekty chlopně a zhoršenému intrakardiálnímu průtoku krve s následným městnavým srdečním selháním.
Komplikace hamartomatózních polypů GI traktu jsou gastrointestinální krvácení, obstrukce a střevní intususcepce (s možným fatálním výsledkem). A velký renální hamartoma může vyvolat prasknutí ledvin.
Hamartoma v mozku může způsobit obstrukční syndrom hydrocefalu.
U hypothalamických a hypofýzach hamartomů může být produkce somatotropního hormonu (růstový hormon) narušena, což vede k rozvoji hypofyzálního nanismu (hypopituitarismus) u dětí. Hypothalamické hamartomy u dětí mohou také vést k epilepsii odolné vůči drogám.
Komplikace epitelu sítnicového pigmentu Hamartoma jsou plné dysfunkce sítnice a/nebo optického nervu, makulárním edémem, neovaskularizací choroidu a oddělením sítnice.
Diagnostika hamartomy
Důležitou součástí diagnózy hamartomů a souvisejících syndromů je sbírka anamneze, včetně rodinné historie.
Laboratorní testy zahrnují krevní testy: obecná klinická; sérové elektrolyty; Profil lymfocytů; hladina vápníku, draslíku, fosfátu a močoviny; a testy jaterních funkcí. Pokud je to možné, provádí se biopsie hmoty jemné jehry aspirace hmoty, protože histologické vyšetření je zásadní v diagnostice a výběru léčebné taktiky.
Instrumentální diagnostika poskytuje vizualizaci hamartomatózní tvorby nádoru a identifikaci jeho přesné lokalizace, pro kterou se používají rentgenová a angiografie, elektroencefalografie (EEG), ultrazvuk (sonografie), CT (počítačová tomografie), PET (tomografie positronu), MRI (magnetická rezonance).
Diferenciální diagnostika
V jakýchkoli abnormálních masách je diferenciální diagnóza velmi důležitá. Tuberkuloma a hamartoma jsou tedy diferencovány; Plicní hamartoma a primární rakovina plic, bronchogenní karcinoid, metastatické onemocnění. Hamartoma mozku by se měl odlišit od kraniofaryngiomu a hypothalamicko-chiasmatického gliomu. A diferenciální diagnóza hamartomu jako vrozeného mesoblastického nefromu zahrnuje Wilmsový nádor (maligní nefroblastom), sarkom čistého buněk ledviny a osifikace ledvin u kojenců.
Kdo kontaktovat?
Léčba hamartomy
Pokud je hamartoma asymptomatický a je náhodně objeven, není nutná žádná léčba, ale je nutné sledovat jeho „chování“ a stav pacienta. V jiných případech je terapie zaměřena na snížení intenzity symptomů a zabránění komplikací. Například v hypothalamickém hamartomu s příznaky předčasné puberty jsou předepsány určité léky, které inhibují uvolňování některých hormonů. Srdeční léky se používají k léčbě příznaků srdečního selhání u pacientů se srdečními hamartomy.
Chirurgické odstranění hamartomů je indikováno pro potvrzení diagnózy a v případě lékařsky neopravitelných intenzivních příznaků.
Například hamartomy plic mohou být resekovány klínovou resekcí a ve závažných případech odstraněním laloku plic (lobektomie). Hamartoma prsu může být také vyříznut, a pokud je velký, může být vyžadována částečná nebo úplná mastektomie.
K odstranění hamartomatózních polypů lze použít stereotaktickou radiofrekvenční termoablaci nebo laserovou ablaci. Používá se také radiochirurgie s vysoce zaměřeným gama paprsky - gama nůž pro hypotalamické hamartomy nebo astrocytární hamartomy.
Prevence
Jedinou metodou prevence vývoje hamartomů lze zvážit genetický screening z budoucích rodičů dítěte.
Předpověď
Celková prognóza této vrozené anomálie závisí na lokalizaci a velikosti novotvaru, jakož i na komorbiditách a obecném zdraví pacienta.