
Apera syndrom
Naposledy posuzováno: 23.04.2024

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.

 [
[Příčiny aperův syndrom
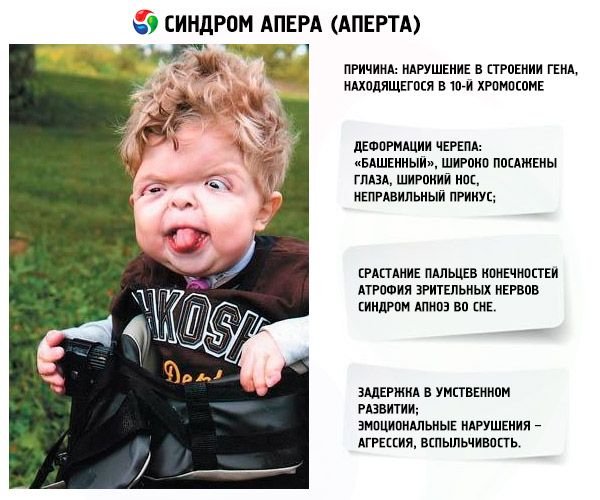
Vývoj syndromu je vyvolán narušením struktury genu lokalizovaného v 10. Chromozomu. Tento gen je zodpovědný za proces oddělování prstů a navíc za včasné uzavření kraniálních stehů.
Kromě toho způsobí, že vzhled patologie uvažovaného přesunuty do březosti infekčních onemocnění (jako je tuberkulóza nebo zarděnky, syfilis nebo chřipky a meningitida), matka rentgenovým zářením. Nejčastěji se podobný syndrom vyskytuje u dětí narozených starším rodičům.
Patogeneze
Syndrom Apera se přenáší dědičnou cestou. Jeho typ je autozomálně dominantní (tj. V rodině, ve které je jeden z rodičů nemocný, pravděpodobnost dítěte s touto chorobou je 50-100%).
Jedinečný růstový faktor fibroblastového receptoru 2 (FGFR2) způsobený mutací vede ke zvýšení počtu progenitorových buněk, které se vyvíjejí podél osteogenní dráhy. Nakonec to vede ke zvýšené tvorbě subperiosteální kostní matrice a předčasné osifikaci kraniálních stehů během vývoje plodu. Pořadí a rychlost tavení stehu určuje stupeň deformace a postižení. Po růstu materiálu na sešívání se růst jiných tkání kolmých na tento steh stává omezeným a sloučené kosti působí jako jediný kostní skelet.
První genetický důkaz, že Syndaktylie na Apertův syndrom je důsledkem keratinocytový růstový defekt receptoru faktoru (KGFR) se pozorování korelace mezi expresí fibroblastů v KGFR a závažnosti Syndaktylie.
Amblyopie a šilhání je častější u pacientů s mutantním FGFR2 Ser252Trp a atrofie zrakového nervu je častější u pacientů s mutantním FGFR2 Pro253Arg. U pacientů s FGR2 Ser252Trp mají mutace významně vyšší prevalenci poškození zraku ve srovnání s pacienty s mutací FGFR2 Pro253Arg.
Symptomy aperův syndrom
Jednotlivé projevy nemoci jsou jasně viditelné i při narození dítěte, protože se rozvíjejí dokonce iv matčině lůně. Mezi hlavní symptomy syndromu patří:
- Lebka je deformovaná - natáhla se na výšku a stala se "věží"; Kromě toho jsou oči široce vysázeny a mírně vyčnívající (protože velikost očních drah klesá); se nos rozšiřuje a vzniká nesprávný skus (horní zuby vyčnívají nadměrně);
- Prsty končetin jsou zcela roztavené (většinou neoznačené a také uprostřed s indexem) a vypadají jako kožní membrána nebo úplná fúze kostí; Navíc mohou růst další prsty;
- Zpoždění v mentálním vývoji (vůbec ne);
- Optické nervy jsou atrofované, což vede k zhoršení zrakové ostrosti (v některých případech se vyvine úplný nedostatek vidění);
- Zvýšený nitrolební tlak, který je důsledkem příliš brzkého přelití kraniálních stehů, se projevuje jako bolest hlavy a zvracení s nevolností;
- Protože horní čelist zůstává nedostatečně rozvinutá, dochází k problémům s dýcháním;
- Syndrom spánkové apnoe je častý.
- Emocionální projevy - agrese, nedostatek zdrženlivosti, silná nálada.
Komplikace a důsledky
Diagnostika aperův syndrom
Pro diagnózu jsou zapotřebí následující opatření:
- Lékař by měl analyzovat stížnosti pacienta i lékařskou anamnézu. Je třeba zjistit, zda v rodině došlo k rozvoji takové patologie;
- Neurologické vyšetření k posouzení tvaru lebky a intelektuálního vývoje pacienta (speciální dotazníky, stejně jako rozhovor);
- Vyšetření fundusu k identifikaci přítomnosti příznaků zvýšeného intrakraniálního tlaku (edém DZN, stejně jako rozmazání jeho okrajů);
- Pro posouzení stavu lebky se provádí jeho radiografie;
- Počítač, stejně jako magnetická rezonance hlava layerwise zkoumat mozkové strukturu lebky, k určení přítomnosti příznaků předčasné fúze kraniálních stehy, ale navíc je hydrocefalus (v důsledku zvýšeného nitrolebního tlaku akumuluje přebytečné kapaliny (tento mozkomíšního moku, který podporuje proces výměny látek, jakož i mozku výživy));
- X-ray nohou s kartáčem, aby zjistil příčinu, kvůli níž došlo k fúzi prstů (to je důležité pro plánování následného chirurgického zákroku);
- Může být předepsána konzultace s lékařem a neurochirurgem.
Analýzy
Genetická analýza častých mutací vyskytujících se v genu typu FGFR2 se provádí.
Diferenciální diagnostika
Tento syndrom odlište od ostatních genetických patologií, u kterých je pozorována kraniosynostóza. Jedná se o nemoci, jako jsou Pfeiffer, Cruson a Sethra-Chotzen a Carpenter syndromy. Pro vyloučení těchto anomálií se používají molekulárně-genetické metody testování.
Kdo kontaktovat?
Léčba aperův syndrom
Provádění operace je považováno za jediný účinný způsob léčby syndromu Apera - pomáhá napravit individuální fyzické vady a také koriguje zpomalení v duševním vývoji.
V průběhu tohoto postupu je uzavírání koronálního stehu prováděno, aby se zabránilo možnému traumatu mozku. Nejběžnější metodou je kraniofaciální rozptýlení, ve kterém se provádí gradace lebky. Pro odstranění jednotlivých defektů na obličeji je provedena ortodontická a / nebo ortogonální operace.
Navíc pacienti chirurgicky odstraní fúzi prstů.