Lékařský expert článku

Nové publikace
Treacherův Collinsův syndrom
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.

Intrauterinní poruchy v procesech vývoje kostí způsobují závažné kraniofaciální deformity a jednou z variant takové patologie je syndrom Treacher-Collins (TCS) nebo mandibulofasciální, tj. maxilofaciální dysostóza.
Kód onemocnění podle MKN 10: třída XVII (vrozené anomálie, deformace a chromozomální poruchy), Q75.4 - mandibulofaciální dysostóza.
Příčiny Treacherův Collinsův syndrom
Tento syndrom byl pojmenován po vynikajícím britském oftalmologovi Edwardu Treacheru Collinsovi, který popsal hlavní rysy patologie před více než sto lety. Evropští lékaři však tento typ anomálie obličejových a čelistních kostí častěji nazývají Franceschettiho chorobou nebo syndromem - na základě rozsáhlého výzkumu švýcarského oftalmologa Adolfa Franceschettiho, který v polovině minulého století zavedl termín „mandibulofasciální dysostóza“. V lékařských kruzích se používá také název Franceschettiho-Collinsův syndrom.
Treacher Collinsův syndrom je způsoben mutacemi v genu TCOF1 (na chromozomálním lokusu 5q31.3-33.3), který kóduje nukleolární fosfoprotein zodpovědný za tvorbu kraniofaciální části lidského embrya. V důsledku předčasného poklesu množství tohoto proteinu dochází k narušení biogeneze a funkcí rRNA. Podle genetiků z výzkumného programu Human Genome tyto procesy vedou ke snížení proliferace embryonálních buněk neurální lišty - hřebene podél neurální drážky, která se během embryonálního vývoje uzavírá do neurální trubice.
K tvorbě obličejových tkání dochází v důsledku transformace a diferenciace buněk horní (hlavové) části neurální lišty, které migrují podél neurální trubice do oblasti prvního a druhého žaberního oblouku embrya. A nedostatek těchto buněk způsobuje kraniofaciální deformace. Kritické období pro výskyt anomálií je od 18. do 28. dne po oplodnění. Po dokončení migrace buněk neurální lišty (ve čtvrtém týdnu těhotenství) se tvoří téměř všechny volné mezenchymální tkáně v obličejové oblasti, které se později (od 5. do 8. týdne) diferencují na kosterní a pojivové tkáně všech částí obličeje, krku, hrtanu, ucha (včetně vnitřního ucha) a budoucích zubů.
Patogeneze
Patogeneze syndromu Treacher-Collins je často familiární a anomálie se dědí autozomálně dominantním způsobem, ačkoli existují případy autozomálně recesivního přenosu vady (s mutacemi v jiných genech, zejména POLR1C a POLR1D). Nejnepředvídatelnější věcí u maxilofaciální dysostózy je, že mutace je děděna dětmi pouze ve 40-48 % případů. To znamená, že u 52-60 % pacientů nejsou příčiny syndromu Treacher-Collins spojeny s přítomností anomálie v rodině a předpokládá se, že patologie se vyskytuje v důsledku sporadických genových mutací de novo. Nové mutace jsou s největší pravděpodobností důsledkem teratogenních účinků na plod během těhotenství.
Mezi teratogenní příčiny tohoto syndromu odborníci uvádějí velké dávky ethanolu (etylalkoholu), záření, cigaretový kouř, cytomegavirus a toxoplazma, stejně jako herbicidy na bázi glyfosátu (Roundal, Glyfor, Tornado atd.). Seznam iatrogenních faktorů zahrnuje léky na akné a seboreu s kyselinou 13-cis-retinovou (Isotretinoin, Accutane); antikonvulzivum fenytoin (Dilantin, Epanutin); psychotropní léky diazepam, valium, relanium, seduxen.
Symptomy Treacherův Collinsův syndrom
Klinické příznaky mandibulofasciální dysostózy a stupeň jejich projevu závisí z velké části na charakteristikách projevu genových mutací. A první příznaky této anomálie jsou ve většině případů viditelné u dítěte ihned po narození: obličej se syndromem Treacher-Collins má charakteristický vzhled. Morfologické anomálie jsou navíc obvykle bilaterální a symetrické.
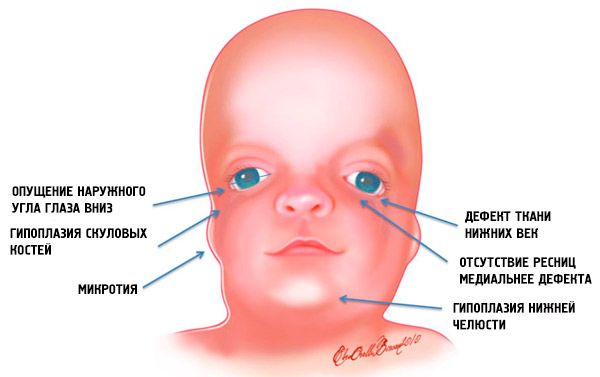
Nejzřetelnější příznaky syndromu Treacher-Collins jsou:
- nedostatečný vývoj (hypoplazie) obličejových kostí lebky: zygomatické, zygomatické výběžky čelní kosti, laterální pterygoidní destičky, paranazální dutiny, dolní čelist a výběžky kostních epifýz (kondyly);
- nedostatečný vývoj kostí dolní čelisti (mikrognatie) a tupější úhel dolní čelisti než obvykle;
- nos má normální velikost, ale zdá se velký kvůli hypoplazii nadočnicových oblouků a nedostatečnému vývoji nebo absenci jařmových oblouků v temporální oblasti;
- oční štěrbiny směřují dolů, tj. tvar očí je abnormální, s vnějšími koutky pokleslými dolů;
- defekty dolních víček (kolobom) a částečná absence řas na nich;
- nepravidelně tvarované boltce s širokou škálou odchylek, včetně jejich umístění v rohu dolní čelisti, absence lalůčků, slepých píštělí mezi tragem ucha a rohem úst atd.;
- zúžení nebo uzavření (atrézie) zevního zvukovodu a anomálie středoušních kůstek;
- absence nebo hypoplazie příušních slinných žláz;
- hypoplazie hltanu (zúžení hltanu a dýchacích cest);
- nesrůstání tvrdého patra (rozštěp patra), stejně jako absence, zkrácení nebo nehybnost měkkého patra.
Takové anatomické anomálie mají ve všech případech komplikace. Jedná se o funkční poruchy sluchu ve formě převodní ztráty sluchu nebo úplné hluchoty; zrakové postižení v důsledku nesprávného vývoje očních bulv; vady patra způsobují potíže s krmením a polykáním. Existují poruchy zubního skusu (malokluze) spojené s vadami čelistí, které následně způsobují problémy se žvýkáním a artikulací. Patologie měkkého patra vysvětlují nosový hlas.
Komplikace a důsledky
Důsledky maxilofaciálních anomálií u syndromu Treacher Collins spočívají v tom, že při narození jsou intelektuální schopnosti dítěte normální, ale v důsledku sluchových vad a dalších poruch je pozorována sekundární mentální retardace.
Děti s takovými vadami navíc akutně pociťují svou méněcennost a trpí, což negativně ovlivňuje jejich nervový systém a psychiku.
Diagnostika Treacherův Collinsův syndrom
Postnatální diagnóza Treacher-Collinsova syndromu je v zásadě založena na klinických příznacích. Kraniofaciální dysostóza je snadno identifikovatelná, pokud je syndrom plně expresivní, ale pokud jsou přítomny minimálně vyjádřené patologické příznaky, mohou nastat problémy se stanovením správné diagnózy.
V tomto případě je třeba věnovat zvláštní pozornost posouzení všech funkcí spojených s anomáliemi, zejména těch, které ovlivňují dýchání (kvůli riziku spánkové apnoe). Měla by být také posouzena a monitorována účinnost krmení a saturace hemoglobinu kyslíkem.
Později, 5.–6. den po porodu, bude nutné zjistit rozsah poškození sluchu pomocí audiologického vyšetření, které by mělo být provedeno v porodnici.
Je předepsáno vyšetření, během kterého se provádí instrumentální diagnostika fluoroskopií kraniofaciální dysmorfologie; pantomografie (panoramatický rentgen kostních struktur obličejové lebky); kompletní kraniální počítačová tomografie v různých projekcích; CT nebo MRI mozku pro určení stavu vnitřního zvukovodu.
Nejranější – prenatální – diagnostika maxilofaciálních anomálií při přítomnosti Treacher Collinsova syndromu v rodinné anamnéze je možná biopsií choriových klků v 10.–11. týdnu těhotenství (zákrok ohrožuje potrat a infekci dělohy).
Krevní testy se odebírají také členům rodiny; v 16.–17. týdnu těhotenství se analyzuje plodová voda (transabdominální amniocentéza); v 18.–20. týdnu těhotenství se provádí fetoskopie a odebírá se krev z fetálních cév placenty.
Nejčastěji se však ultrazvuk používá při prenatální diagnostice tohoto syndromu u plodu (ve 20.–24. týdnu těhotenství).
Jaké testy jsou potřeba?
Diferenciální diagnostika
Stejné metody používají specialisté, když je potřeba diferenciální diagnostika k rozpoznání mírného syndromu Treacher-Collins a jeho odlišení od jiných vrozených anomálií kraniofaciálních kostí, zejména: Apertova, Crouzonova, Nagerova, Petersova-Hewelsova, Hellermannova-Stephova syndromu, stejně jako hemifaciální mikrosomie (Goldenharův syndrom), hypertelorismu, předčasného srůstu lebečních švů (kraniosynostóza) nebo zhoršeného srůstu obličejových kostí (kraniosynostóza).
Léčba Treacherův Collinsův syndrom
Stejně jako ve všech případech geneticky podmíněných vrozených vad je léčba těžkých forem syndromu Treacher-Collins výhradně paliativní, protože pro takové patologie jednoduše neexistují žádné terapeutické metody. Spektrum a stupeň deformací u tohoto syndromu jsou rozsáhlé, a proto má i povaha a intenzita lékařského zásahu mnoho možností.
Sluchadla se používají ke korekci a zlepšení sluchu a sezení logopedie ke zlepšení řeči.
Chirurgické zákroky jsou nutné v raném věku u závažných případů zúžení dýchacích cest (provádí se tracheostomie) a hrtanu (provádí se gastrostomie pro podávání výživy). Může být nutná i chirurgická korekce patra.
Operace prodloužení mandibuly se provádějí ve věku 2-3 let nebo později. Rekonstrukce měkkých tkání zahrnuje korekci colobomu dolního víčka a plastickou chirurgii ušních boltců.
Předpověď
Jaká je prognóza této patologie? Záleží na stupni deformace a intenzitě symptomů. Treacher-Collinsův syndrom je celoživotní diagnóza.
 [ 25 ]
[ 25 ]