Lékařský expert článku

Nové publikace
T-buněčné lymfomy kůže
Last reviewed: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
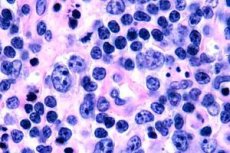
Nejčastěji jsou T-buněčné lymfomy registrovány u starších lidí, i když ojedinělé případy onemocnění jsou zaznamenány i u dětí. Muži jsou nemocní dvakrát častěji než ženy. T-buněčné lymfomy mají epidermotropní povahu.
Příčiny T-buněčné lymfomy kůže
Příčiny a patogeneze kožních T-buněčných lymfomů nejsou zcela objasněny. V současné době většina vědců považuje virus lidské T-buněčné leukémie typu 1 (HTLV-1) I za hlavní etiologický faktor iniciující vývoj maligních T-buněčných lymfomů kůže. Spolu s tím se diskutuje role dalších virů ve vývoji T-buněčného lymfomu: virus Epstein-Barrové, herpes simplex typ 6. U pacientů s T-buněčným lymfomem se viry nacházejí v kůži, periferní krvi a Langerhansových buňkách. Protilátky proti HTLV-I jsou detekovány u mnoha pacientů s mycosis fungoides.
Důležité místo v patogenezi T-buněčných lymfomů hrají imunopatologické procesy v kůži, z nichž hlavní je nekontrolovaná proliferace klonálních lymfocytů.
Cytokiny produkované lymfocyty, epiteliálními buňkami a buňkami makrofágového systému mají prozánětlivé a proliferativní účinky (IL-1, zodpovědný za diferenciaci lymfocytů; IL-2 - T-buněčný růstový faktor; IL-4 a IL-5, zvyšující příliv eosinofilů do léze a jejich aktivaci atd.). V důsledku přílivu T-lymfocytů do léze se tvoří Pautrierovy mikroabscesy. Současně se zvýšením proliferace lymfocytů je potlačena aktivita protinádorových obranných buněk: přirozených zabíječů, lymfocytotoxických lymfocytů, dendritických buněk, zejména Langerhansových buněk, a také cytokinů (IL-7, IL-15 atd.) - inhibitorů růstu nádoru. Nelze vyloučit roli dědičných faktorů. Přítomnost familiárních případů, častá detekce některých antigenů histokompatibility (HLA B-5 a HLA B-35 - u vysoce maligních kožních lymfomů, HLA A-10 - u méně agresivních lymfomů, HLA B-8 - u erytrodermické formy mycosis fungoides) potvrzují dědičnou povahu dermatózy.
Klinická pozorování naznačují možnou transformaci dlouhodobých chronických dermatóz (neurodermatitida, atopická dermatitida, psoriáza atd.) do mycosis fungoides. Klíčovým faktorem je dlouhodobá perzistence lymfocytů v ložisku zánětu, které narušují imunitní dohled a podporují vznik klonu maligních lymfocytů, a tím i rozvoj maligního proliferativního procesu.
Vliv fyzikálních faktorů na tělo, jako je sluneční záření, ionizující záření a chemické látky, může vést ke vzniku klonu „genotraumatických“ lymfocytů, které mají mutagenní účinek na lymfoidní buňky a rozvoj malignity lymfocytů.
T-buněčné lymfomy lze proto považovat za multifaktoriální onemocnění, které začíná aktivací lymfocytů pod vlivem různých karcinogenních, „genotraumatizujících“ faktorů a vznikem dominantního klonu T-buněk. Závažnost poruchy imunitního dohledu, klon maligních lymfocytů, určuje klinické projevy (skvrnité, plakové nebo nádorové elementy) T-buněčných lymfomů.
Patogeneze
V rané fázi mycosis fungoides se pozoruje akantóza s širokými výběžky, hyperplazie a zhutnění bazálních keratinocytů, vakuolární degenerace některých bazálních buněk, atypické mitózy v různých vrstvách epidermis, epidermotropismus infiltrátu s pronikáním lymfocytů do epidermis. V dermis se kolem cév objevují malé infiltráty, sestávající z jednotlivých mononukleárních buněk s hyperchromními jádry - "mykotické" buňky. Ve druhé fázi se pozoruje zvýšení závažnosti dermálního infiltrátu a epidermotropismus buněk infiltrátu, v důsledku čehož maligní lymfocyty pronikají do epidermis a tvoří shluky ve formě Potrierových mikroabscesů. Ve třetí, nádorové fázi se pozoruje masivní akantóza a drobná atrofie epidermis, stejně jako zvýšená infiltrace epidermis nádorovými lymfocyty, které tvoří mnohočetné Potrierovy mikroabscesy. Masivní infiltrát se nachází v celé tloušťce dermis a pokrývá část hypodermis. Pozorují se blastové formy lymfocytů.
Kožní velkoanaplastický T-buněčný lymfom
Je reprezentována skupinou lymfoproliferativních procesů charakterizovaných přítomností proliferátů z atypických klonálních velkých anaplastických CD30+ T buněk. Zpravidla se sekundárně vyvíjí v nádorovém stádiu mycosis fungoides nebo u Sezaryho syndromu, ale může se vyvinout samostatně nebo s diseminací systémových lymfomů tohoto typu. Klinicky tyto lymfomy odpovídají tzv. dekapitované formě mycosis fungoides ve formě jednotlivých nebo vícečetných uzlin, obvykle seskupených.
Histologicky proliferát zabírá téměř celou dermis s epidermotropismem nebo bez něj v případě epidermální atrofie.
Cytologicky se nádorové buňky mohou lišit velikostí a tvarem. Na základě těchto vlastností se rozlišuje mezi středně a velkobuněčným pleomorfním T-buněčným lymfomem s jádry různých nepravidelných konfigurací - spletitými, vícelaločnými, s hustým chromatinem, dobře definovaným jadérkem a poměrně hojnou cytoplazmou; imunoblastickým - s velkými kulatými nebo oválnými jádry s čirou karyoplazmou a jedním centrálně umístěným jadérkem; anaplastickým - s ošklivými velmi velkými buňkami s jádry nepravidelné konfigurace a hojnou cytoplazmou. Fenotypicky celá tato skupina patří k T-helper lymfomům a může být CD30+ nebo CD30-.
R. Willemze a kol. (1994) prokázali, že průběh CD30+ lymfomu je příznivější. Genotypicky je detekována klonální přeskupení receptoru T-lymfocytů.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Symptomy T-buněčné lymfomy kůže
Nejčastějším onemocněním ve skupině T-buněčných lymfomů kůže je mycosis fungoides, která představuje asi 70 % případů. Existují tři klinické formy onemocnění: klasická, erytrodermická a deheaded. T-buněčné lymfomy se vyznačují polymorfismem vyrážek ve formě skvrn, plaků, nádorů.
Erytrodermická forma mycosis fungoides obvykle začíná nekontrolovatelným svěděním, otokem, univerzální hyperémií, výskytem erytematózně-dlaždicových lézí na kůži trupu a končetin, které mají tendenci splývat a rozvíjet erytrodermii během 1-2 měsíců. Téměř všichni pacienti mají palmárně-plantární hyperkeratózu a difúzní ztenčení vlasů po celé kůži. Všechny skupiny lymfatických uzlin jsou značně zvětšené. Zvětšené tříselné, femorální, axilární a loketní lymfatické uzliny jsou palpovány jako "balíčky" husté elastické konzistence, nesrostlé s okolními tkáněmi, bezbolestné. Celkový stav se prudce zhoršuje: objevuje se horečka s tělesnou teplotou až 38-39 °C, noční pocení, slabost a úbytek hmotnosti. V současné době je Sezaryho syndrom mnoha dermatology považován za nejvzácnější leukemickou variantu erytrodermické formy mycosis fungoides,
Na lymfocytogramech je zaznamenána výrazná leukocytóza - Sezaryho buňky. Sezaryho buňky jsou maligní T-helpery, jejichž jádra mají složený cerebriformní povrch s hlubokými invaginacemi jaderné membrány. Fatální výsledek je zaznamenán po 2-5 letech, jehož častou příčinou je kardiovaskulární patologie a intoxikace.
Dekapitovaná forma mycosis fungoides se vyznačuje rychlým vývojem nádorovitých lézí na zdánlivě zdravé kůži bez předchozí dlouhodobé tvorby plaků. Tato forma se vyznačuje vysokým stupněm malignity, který je považován za projev lymfosarkomu. Fatální výsledek je pozorován do jednoho roku.
Etapy
Klasická forma mycosis fungoides se vyznačuje třemi vývojovými stádii: erytematózně-dlaždicovou, plakovou a tumorovou.
První stádium připomíná klinický obraz některých benigních zánětlivých dermatóz - ekzému, seboroické dermatitidy, plakové parapsoriázy. V tomto stádiu onemocnění se pozorují skvrny různých velikostí, intenzivně růžové, růžovočervené s fialovým odstínem, kulatých nebo oválných obrysů, s relativně jasnými hranicemi, povrchovým otrubovitým nebo jemně lamelovým olupováním. Prvky se často nacházejí na různých oblastech kůže, nejčastěji na trupu a obličeji. Postupně se jejich počet zvyšuje. Postupem času může proces nabýt charakteru erytrodermie (erytrodermické stádium). Vyrážka může existovat roky nebo spontánně vymizet. Na rozdíl od benigních zánětlivých dermatóz jsou prvky vyrážky a svědění v tomto stádiu rezistentní na terapii.
Infiltrativně-plakové stádium se vyvíjí v průběhu několika let. Na místě dříve existujících skvrnitých vyrážek se objevují plaky kulatého nebo nepravidelného obrysu, intenzivně fialové barvy, jasně ohraničené od zdravé kůže, husté, s šupinatým povrchem. Jejich konzistence připomíná „tlustý karton“. Některé z nich spontánně vymizí a zanechávají oblasti tmavě hnědé hyperpigmentace a/nebo atrofie (poikiloderma). Svědění je v tomto stádiu ještě intenzivnější a bolestivější, pozoruje se horečka a úbytek hmotnosti. V tomto stádiu může být pozorována lymfadenopatie.
Ve třetím, nádorovém stádiu, se objevují bezbolestné nádory husté, elastické konzistence, žlutočervené barvy, vyvíjející se z plaků nebo vznikající na zdánlivě zdravé kůži. Tvar nádorů je kulovitý nebo zploštělý, často připomínající houbovitý klobouk. Nádory se mohou objevit kdekoli. Jejich počet se značně liší od jednotlivých až po desítky, velikosti - od 1 do 20 cm v průměru. Při rozpadu dlouhodobě existujících nádorů se tvoří vředy s nerovnými okraji a hlubokým dnem, zasahující do fascie nebo kosti. Nejčastěji jsou postiženy lymfatické uzliny, slezina, játra a plíce. Celkový stav se zhoršuje, objevují se a stupňují se příznaky intoxikace, rozvíjí se slabost. Průměrná délka života pacientů s klasickou formou mycosis fungoides od okamžiku diagnózy je 5 až 10 let. Úmrtnost je obvykle pozorována z interkurentních onemocnění: pneumonie, kardiovaskulární selhání, amyloidóza. Subjektivně se pociťuje svědění a při rozpadu nádorů bolest v postižených oblastech.
Co je třeba zkoumat?
Jak zkoušet?
Léčba T-buněčné lymfomy kůže
V erytematózně-dlaždicovém stádiu pacienti nepotřebují protinádorovou terapii; předepisují se jim lokální kortikosteroidy (prednisolon, betamethason, deriváty dexamethasonu), interferon alfa (3 miliony IU denně, poté 3krát týdně po dobu 3-6 měsíců v závislosti na klinických projevech nebo účinnosti léčby), interferon gama (100 000 IU denně po dobu 10 dnů, cyklus se opakuje 12-3krát s 10denní přestávkou), PUVA terapie nebo Re-PUVA terapie. Účinnost PUVA terapie je založena na selektivní tvorbě kovalentních zesítění psoralenů s DNA v proliferujících T-helper buňkách, což inhibuje jejich dělení. Ve druhém stádiu se kromě výše uvedených látek používají systémové kortikosteroidy (30-40 mg prednisolonu denně po dobu 1,5-2 měsíců) a cytostatika (prospedin 100 mg denně denně, celkem 4-5 injekcí). Kombinace interferonů s jinými metodami terapie má výraznější terapeutický účinek (interferony + PUVA, interferony + cytostatika, interferony + aromatické retinoidy).
V nádorovém stádiu je hlavní metodou polychemoterapie. Používá se kombinace vinkristinu (0,5-1 mg intravenózně jednou denně, celkem 4-5 injekcí) s prednisolonem (40-60 mg denně perorálně během chemoterapie), prospidinem (100 mg denně, celkem 3 g) a interferony. Doporučuje se fotodynamická terapie, terapie elektronovým paprskem a fotoferéza (mimotělní fotochemoterapie).