Lékařský expert článku

Nové publikace
Usherův syndrom
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Usherův syndrom je dědičné onemocnění, které se projevuje úplnou hluchotou od narození a také progresivní slepotou s věkem. Ztráta zraku je spojena s retinitis pigmentosa, procesem pigmentové degenerace sítnice. Mnoho lidí s Usherovým syndromem má také vážné problémy s rovnováhou.
Epidemiologie
Díky výzkumu bylo možné zjistit, že Usherův syndrom postihuje přibližně 8 % vyšetřených hluchoněmých dětí (testy byly provedeny ve speciálních zařízeních pro hluchoněmé osoby). Pigmentární retinitida byla pozorována u 6–10 % pacientů trpících vrozenou hluchotou, která je zase pozorována u přibližně 30 % lidí s pigmentovým onemocněním sítnice.
Předpokládá se, že toto onemocnění se na celém světě projevuje přibližně u 3–10 lidí ze 100 tisíc. Může se vyskytovat stejnou měrou u žen i mužů. Tímto syndromem trpí přibližně 5–6 % světové populace. Asi 10 % všech případů těžké dětské hluchoty je způsobeno Usherovým syndromem I. i II. typu.
Ve Spojených státech jsou nejčastějšími typy 1 a 2. Dohromady představují přibližně 90 až 95 procent všech případů Usherova syndromu u dětí.
Příčiny Usherův syndrom
Usherův syndrom typu I, II a III má autozomálně recesivní příčinu, zatímco typ IV je považován za poruchu chromozomu X. Příčiny slepoty a hluchoty, které se u tohoto syndromu vyskytují, dosud nebyly dostatečně prozkoumány. Předpokládá se, že lidé s tímto onemocněním jsou přecitlivělí na složky, které mohou poškodit strukturu DNA. Kromě toho může být toto onemocnění spojeno s poruchami imunitního systému, ale v tomto případě neexistuje přesný obraz tohoto procesu.
V roce 1989 byly u pacientů s onemocněním typu II poprvé identifikovány chromozomální abnormality, což by v budoucnu mohlo vést k izolaci genů, které tento syndrom způsobují. Je také možné identifikovat tyto geny u nositelů a vyvinout speciální prenatální genetické testy.
 [ 8 ]
[ 8 ]
Rizikové faktory
Syndrom se dědí, pokud jsou postiženi oba rodiče, tj. dědí se recesivním typem. Dítě může zdědit onemocnění i v případě, že jeho rodiče jsou nositeli genu. Pokud mají oba budoucí rodiče tento gen, pak je pravděpodobnost, že se narodí dítě s tímto syndromem, 1:4. Osoba, která má pouze jeden gen pro tento syndrom, je považována za nositele, ale nemá příznaky onemocnění. V dnešní době zatím není možné určit, zda daná osoba má gen pro toto onemocnění.
Pokud se dítě narodí rodičům, z nichž jeden takový gen nemá, pak je pravděpodobnost, že syndrom zdědí, velmi nízká, ale rozhodně bude nositelem.
Symptomy Usherův syndrom
Mezi příznaky Usherova syndromu patří ztráta sluchu a abnormální hromadění pigmentovaných buněk v očních strukturách. U pacienta se následně rozvíjí degenerace sítnice, která způsobuje zhoršení zraku a v nejzávažnějších případech až ztrátu zraku.
Senzorineurální ztráta sluchu může být mírná nebo úplná a obvykle neprogreduje od narození. Pigmentové onemocnění sítnice se však může začít rozvíjet v dětství nebo později. Výsledky testů ukázaly, že centrální zraková ostrost může být zachována po mnoho let, a to i při zhoršení periferního vidění (stav nazývaný „tunelové vidění“).
Toto jsou hlavní projevy onemocnění, které mohou být někdy doplněny dalšími poruchami, jako jsou psychózy a další duševní poruchy, problémy s vnitřním uchem a/nebo šedý zákal.
Formuláře
Během výzkumu byly identifikovány 3 typy tohoto onemocnění a také 4. forma, která je poměrně vzácná.
Typ I onemocnění je charakterizován vrozenou úplnou hluchotou a také poruchou rovnováhy. Takové děti často začínají chodit až ve věku 1,5 roku. Zhoršování zraku obvykle začíná ve věku 10 let a konečný rozvoj stavu šerosleposti začíná ve věku 20 let. U dětí s tímto typem onemocnění se může vyvinout progresivní zhoršování periferního vidění.
U onemocnění typu II je pozorována středně těžká nebo vrozená hluchota. V tomto případě se často již zhoršení částečné hluchoty nedostavuje. Pigmentární retinitida se začíná rozvíjet kolem konce adolescence nebo po 20 letech. Vývoj šerosleposti obvykle začíná ve 29–31 letech. Zhoršení zrakové ostrosti u patologie typu II obecně postupuje o něco pomaleji než u typu I.
Typ III onemocnění je charakterizován progresivní ztrátou sluchu, obvykle začínající během puberty, a také postupným rozvojem retinitis pigmentosa ve stejném období (o něco později než ztráta sluchu), která se může stát faktorem při rozvoji progresivní slepoty.
Projevy patologie typu IV se vyskytují hlavně u mužů. V tomto případě jsou také pozorovány progresivní poruchy a ztráta sluchu a zraku. Tato forma je velmi vzácná a obvykle má X-chromozomální povahu.
Diagnostika Usherův syndrom
Diagnóza Usherova syndromu se stanoví na základě pozorované kombinace náhlé hluchoty a progresivní ztráty zraku u pacienta.
Testy
K detekci mutace může být nařízen speciální genetický test.
Bylo nalezeno jedenáct genetických lokusů, které mohou způsobit rozvoj Usherova syndromu, a bylo identifikováno devět genů, které jsou definitivní příčinou této poruchy:
- Typ 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Typ 2: ush2a, VLGR1, WHRN.
- Usherův syndrom typu 3: USH3A.
Vědci z NIDCD spolu s kolegy z univerzit v New Yorku a Izraeli identifikovali mutaci nazvanou R245X v genu Pcdh15, která je zodpovědná za velké procento Usherova syndromu typu 1 v židovské populaci.
Chcete-li se dozvědět více o laboratořích, které provádějí klinické studie, navštivte stránky https://www.genetests.org a vyhledejte v adresáři laboratoří výraz „Usherův syndrom“.
Chcete-li se dozvědět o existujících klinických studiích, které zahrnují genetické testování Usherova syndromu, navštivte stránky https://www.clinicaltrials.gov a vyhledejte „Usherův syndrom“ nebo „genetické testování Usherova syndromu“.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentální diagnostika
Existuje několik metod instrumentální diagnostiky:
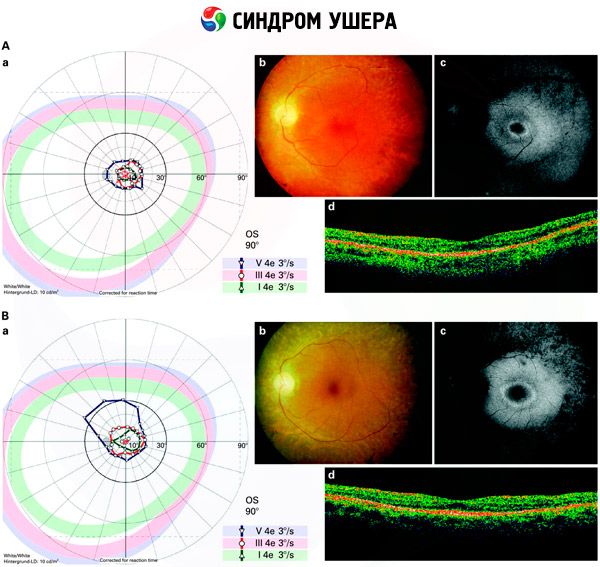
- Vyšetření fundusu za účelem zjištění přítomnosti pigmentových skvrn na sítnici a také zúžení sítnicových cév;
- Elektroretinogram, který umožňuje detekci počátečních degenerativních odchylek v oční sítnici. Zobrazuje zánik elektroradiografických drah;
- Elektronystagmogram (ENG) měří mimovolní pohyby očí, které by mohly naznačovat přítomnost nerovnováhy.
- Audiometrie, která se používá k určení přítomnosti hluchoty a její závažnosti.
Diferenciální diagnostika
Usherův syndrom je nutné odlišit od některých podobných poruch.
Hallgrenův syndrom, který se vyznačuje vrozenou ztrátou sluchu a progresivní ztrátou zraku (vyvíjí se také katarakta a nystagmus). Mezi další příznaky patří ataxie, psychomotorické poruchy, psychóza a mentální retardace.
Alstromův syndrom, což je dědičné onemocnění, při kterém dochází k degeneraci sítnice, což vede ke ztrátě centrálního vidění. Tento syndrom je spojen s dětskou obezitou. Zároveň se po 10 letech začíná rozvíjet diabetes mellitus a ztráta sluchu.
Zarděnky u těhotné ženy v prvním trimestru mohou způsobit různé abnormality ve vývoji dítěte. Mezi důsledky takové abnormality patří ztráta sluchu, stejně jako (nebo) problémy se zrakem a kromě toho různé vývojové vady.
Kdo kontaktovat?
Léčba Usherův syndrom
V současné době neexistuje lék na Usherův syndrom. Terapie v tomto případě proto spočívá především ve zpomalení procesu ztráty zraku a také v kompenzaci ztráty sluchu. Mezi možné léčebné metody patří:
- Užívání vitaminu A (někteří oftalmologové se domnívají, že vysoké dávky palmitátu vitaminu A mohou zpomalit, ale ne zastavit, progresi retinitis pigmentosa);
- Implantace speciálních elektronických zařízení do uší pacienta (sluchadla, kochleární implantáty).
Oftalmologové doporučují, aby většina dospělých s běžnými formami retinitis pigmentosa denně užívala 15 000 IU (mezinárodních jednotek) palmitátu vitaminu A pod dohledem. Protože lidé s Usherovým syndromem typu 1 nebyli do studie zahrnuti, vysoké dávky vitaminu A se u této skupiny pacientů nedoporučují. Lidé, kteří zvažují užívání vitaminu A, by se měli o této možnosti léčby poradit se svým lékařem. Další doporučení pro tuto možnost léčby zahrnují:
- Změňte svůj jídelníček a zařaďte do něj potraviny s vysokým obsahem vitamínu A.
- Ženy plánující těhotenství by měly přestat užívat vysoké dávky vitaminu A tři měsíce před plánovaným početím kvůli zvýšenému riziku vrozených vad.
- Těhotné ženy by měly přestat užívat vysoké dávky vitaminu A kvůli zvýšenému riziku vrozených vad.
Důležité je také adaptovat takové dítě na společenský život. To vyžaduje pomoc speciálních pedagogů a psychologů. V případě, že pacient začal pociťovat progresivní ztrátu zraku, měl by se naučit používat znakovou řeč.
Předpověď
Usherův syndrom má nepříznivou prognózu. U většiny pacientů s tímto onemocněním jakéhokoli typu se zorné pole a jeho ostrost začínají zhoršovat v období 20–30 let. V některých případech dochází k úplné bilaterální ztrátě zraku. Ztráta sluchu, která je vždy doprovázena hloupostí, se velmi rychle vyvine v úplnou bilaterální ztrátu sluchu.