Lékařský expert článku

Nové publikace
Subakutní nekrotizující Leahova encefalomyopatie
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
Toto onemocnění bylo poprvé zmíněno v roce 1951. Do dnešního dne bylo popsáno více než 120 případů. Leighova choroba (OMIM 256000) je geneticky heterogenní onemocnění, které se může dědit buď nukleárně (autozomálně recesivně nebo vázaně na X chromozom), nebo mitochondriálně (méně často).
 [
[ Příčiny Leahova syndromu
Základem onemocnění je deficit enzymů zajišťujících produkci energie, zejména v důsledku narušení metabolismu kyseliny pyrohroznové a poruchy transportu elektronů v dýchacím řetězci. Vzniká deficit komplexu pyruvátdehydrogenázy (podjednotka a-E1), pyruvátkarboxylázy, komplexu 1 (NAD-koenzym Q-reduktáza) a komplexu 4 (cytochromoxidáza) dýchacího řetězce.
Bylo zjištěno, že defekty pyruvátkarboxylázy, komplexu 1 (NAD-koenzym Q-reduktáza) a komplexu 4 (cytochromoxidáza) dýchacího řetězce se dědí autozomálně recesivně, defekty komplexu pyruvátdehydrogenázy (podjednotka a-E1) se dědí recesivně vázaným způsobem na chromozom X. V případě bodových mutací mtDNA, které postihují 6. podjednotku ATPázy, je typická mitochondriální dědičnost. Nejčastěji se vyskytuje miscensová mutace spojená s nahrazením thyminu guaninem nebo cytosinem v pozici 8993 mtDNA. Méně častá je mutace v pozici 9176 mtDNA. Vzhledem k tomu, že mutace T8993G je hlavní vadou u NARP syndromu, byly popsány rodiny s těmito dvěma onemocněními. U dětí byla popsána i mutace v mtDNA v pozici 8344, která se vyskytuje u MERRF syndromu.
Předpokládá se, že v případě akumulace mutantní mtDNA ve většině mitochondrií se vyvíjí těžký průběh Leighova syndromu. V mitochondriální genezi tohoto stavu se mutantní mtDNA nachází v 90 % všech mitochondrií. Patogeneze je spojena s porušením tvorby energie v buňkách a rozvojem laktátové acidózy.
Symptomy Leahova syndromu
První příznaky onemocnění se objevují v raném věku (1-3 roky). Jsou však známy případy manifestace onemocnění ve 2 týdnech a v 6-7 letech. Zpočátku se rozvíjejí nespecifické poruchy: opožděný psychomotorický vývoj, snížená chuť k jídlu, epizody zvracení, deficit tělesné hmotnosti. Následně se stupňují neurologické příznaky: svalová hypotonie nebo dystonie s přechodem do hypertonie, ataky myoklonu nebo tonicko-klonických záchvatů, třes končetin, choreoatetóza, poruchy koordinace, snížené šlachové reflexy, letargie, ospalost. Mozková neurodegenerace progresivní. Zvyšují se příznaky pyramidální a extrapyramidové insuficience, polykací funkce je narušena. Často se pozorují změny v orgánech vidění, jako je ptóza, oftalmoplegie, atrofie zrakových nervů, méně často pigmentová degenerace sítnice. Někdy se rozvíjí hypertrofická kardiomyopatie, objevují se epizody tachypnoe.
Vzácně probíhá onemocnění jako akutní encefalopatie. Typičtější je chronický nebo subakutní průběh, který vede k fatálnímu konci několik let po nástupu onemocnění. Při rychlém průběhu (několik týdnů) nastává smrt v důsledku paralýzy dýchacího centra.
Diagnostika Leahova syndromu
Biochemický krevní test odhaluje laktátovou acidózu v důsledku hromadění kyseliny mléčné a pyrohroznové v krvi a mozkomíšním moku, stejně jako zvýšení obsahu alaninu v krvi. Může být také zvýšena hladina ketonových tělísek. V moči je zjištěno zvýšené vylučování organických kyselin: mléčné, fumarové atd. Hladina karnitinu v krvi a tkáních často klesá.
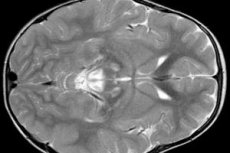
Výsledky EEG odhalují ložiskové známky epileptické aktivity. Data z magnetické rezonance odhalují zvětšení mozkových komor, bilaterální poškození mozku, kalcifikaci bazálních ganglií (nucleus caudatus, putamen, substantia nigra, globus pallidus). Lze také detekovat atrofii mozkových hemisfér a mozkové hmoty.
Morfologické vyšetření odhaluje hrubé změny v mozkové hmotě: symetrická ložiska nekrózy, demyelinizace a houbovité degenerace mozku, zejména středních částí, pontu, bazálních ganglií, thalamu a zrakového nervu. Histologický obraz zahrnuje cystickou degeneraci mozkové tkáně, astrocytární gliózu, odumírání neuronů a zvýšení počtu mitochondrií v buňkách. V kosterních svalech dochází k akumulaci lipidových inkluzí, snížení histochemické reakce na komplexy 1 a 4 dýchacího řetězce, subsarkolemální akumulaci mitochondrií, abnormálním mitochondriím s dezorganizací krist. Fenomén RRF často není detekován.
Jak zkoušet?
Jaké testy jsou potřeba?
Использованная литература