Lékařský expert článku

Nové publikace
Polymyozitida a dermatomyozitida: příčiny, příznaky, diagnostika, léčba
Last reviewed: 05.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.
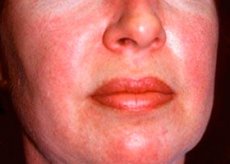
Polymyozitida a dermatomyozitida jsou vzácná systémová revmatická onemocnění charakterizovaná zánětlivými a degenerativními změnami svalů (polymyozitida) nebo svalů a kůže (dermatomyozitida). Nejspecifičtějším kožním projevem je heliotropní vyrážka.
Postižení svalů je symetrické a zahrnuje slabost, určitou citlivost a následnou atrofii proximálních svalů pánevního pletence. Mezi komplikace může patřit viscerální postižení a malignita. Diagnóza je založena na klinické prezentaci a posouzení svalové dysfunkce měřením hladin enzymů, provedením MRI, elektromyografie a svalové biopsie. Léčba zahrnuje glukokortikoidy, někdy v kombinaci s imunosupresivy nebo intravenózními imunoglobuliny.
Ženy onemocní dvakrát častěji než muži. Onemocnění se může objevit v jakémkoli věku, ale nejčastěji se vyskytuje v rozmezí od 40 do 60 let; u dětí - od 5 do 15 let.
 [
[ Co způsobuje dermatomyozitidu a polymyozitidu?
Předpokládá se, že příčinou onemocnění je autoimunitní reakce na svalovou tkáň u geneticky predisponovaných jedinců. Onemocnění je častější u osob s zatíženou rodinnou anamnézou a nositelů určitých HLA antigenů (DR3, DR52, DR56). Možnými spouštěči jsou virová myozitida a maligní novotvary. Existují zprávy o detekci struktur podobných pikornavirům ve svalových buňkách; viry navíc mohou u zvířat vyvolat podobná onemocnění. Souvislost maligních nádorů s dermatomyozitidou (mnohem méně často než s polymyozitidou) naznačuje, že růst nádoru může být také spouštěčem rozvoje onemocnění v důsledku zahájení autoimunitních reakcí na společné antigeny nádoru a svalové tkáně.
Ve stěnách cév kosterních svalů se nacházejí depozita IgM, IgG a třetí složky komplementu; to je zvláště charakteristické pro dermatomyozitidu u dětí. Pacienti s polymyozitidou mohou také vyvinout další autoimunitní procesy.
Patofyziologie dermatomyozitidy a polymyozitidy
Patologické změny zahrnují poškození buněk a atrofii na pozadí zánětu různého stupně závažnosti. Svaly horních a dolních končetin, stejně jako obličej, jsou poškozeny v menší míře než jiné kosterní svaly. Poškození viscerálních svalů hltanu a horní části jícnu, méně často srdce, žaludku nebo střev, může vést k dysfunkci výše uvedených orgánů. Vysoké koncentrace myoglobinu v důsledku rabdomyolýzy mohou způsobit poškození ledvin. Mohou se také vyskytnout zánětlivé změny v kloubech a plicích, zejména u pacientů s protilátkami proti syntetáze.
Příznaky dermatomyozitidy a polymyozitidy
Nástup polymyozitidy může být akutní (zejména u dětí) nebo subakutní (častěji u dospělých). Akutní virová infekce někdy předchází nebo je spouštěčem manifestace onemocnění, jehož nejčastějšími projevy jsou slabost proximálních svalů nebo kožní vyrážky. Bolest je vyjádřena v menší míře než slabost. Může se rozvinout polyartralgie, Raynaudův fenomén, dysfagie, plicní poruchy, celkové příznaky (zvýšená tělesná teplota, snížení tělesné hmotnosti, slabost). Raynaudův fenomén se často vyskytuje u pacientů se souběžnými onemocněními pojivové tkáně.
Svalová slabost může progredovat během několika týdnů nebo měsíců. Pro klinický projev svalové slabosti však musí být postiženo alespoň 50 % svalových vláken (přítomnost svalové slabosti tedy naznačuje progresi myozitidy). Pacienti mohou mít potíže se zvedáním paží nad úroveň ramen, chůzí do schodů nebo vstáváním ze sedu. Kvůli těžké slabosti svalů pánve a ramenního pletence mohou být pacienti upoutáni na invalidní vozík nebo lůžko. Pokud jsou postiženy flexory krku, je nemožné zvednout hlavu z polštáře. Postižení svalů hltanu a horní části jícnu vede k poruchám polykání a regurgitaci. Svaly dolních a horních končetin a obličeje obvykle nejsou postiženy. Mohou se však vyvinout kontraktury končetin.
Kožní vyrážky pozorované u dermatomyozitidy jsou obvykle tmavé barvy a erytematózní. Charakteristický je také periorbitální edém fialové barvy (heliotropní vyrážka). Kožní vyrážky mohou být mírně vyvýšené nad úroveň kůže a hladké nebo šupinaté; lokalizace vyrážek je na čele, krku, ramenou, hrudníku, zádech, předloktí, dolních částech holenních kostí, obočí, v oblasti kolen, mediálních malleolech, dorzálních plochách interfalangeálních a metakarpofalangeálních kloubů, na laterální straně (Gottronův příznak). Možná je hyperémie báze nebo periferie nehtů. Na kůži laterální plochy prstů se může vyvinout deskvamativní dermatitida doprovázená prasklinami. Primární kožní léze často odezní bez následků, ale mohou vést k sekundárním změnám, jako je tmavá pigmentace, atrofie, zjizvení nebo vitiligo. Mohou se vyvinout subkutánní kalcifikace, zejména u dětí.
Přibližně u 30 % pacientů se vyvine polyartralgie nebo polyartritida, často doprovázená otokem a kloubním výpotkem. Závažnost kloubních projevů je však malá. Vyskytují se častěji, pokud jsou u pacientů detekovány protilátky proti Jo-1 nebo jiným syntetázám.
Postižení vnitřních orgánů (s výjimkou hltanu a horní části jícnu) je u polymyozitidy méně časté než u jiných revmatických onemocnění (zejména SLE a systémové sklerózy). Vzácně, zejména u antisyntetázového syndromu, se onemocnění projevuje jako intersticiální pneumonitida (ve formě dušnosti a kašle). Mohou se rozvinout srdeční arytmie a poruchy vedení vzruchů, ale obvykle jsou asymptomatické. Gastrointestinální projevy jsou častější u dětí, které mají také vaskulitidu, a mohou zahrnovat zvracení s krví, melénu a perforaci střeva.
Kde to bolí?
Klasifikace polymyozitidy
Existuje 5 typů polymyozitidy.
- Primární idiopatická polymyozitida, která se může objevit v jakémkoli věku, nepostihuje kůži.
- Primární idiopatická dermatomyozitida je podobná primární idiopatické polymyozitidě, ale postihuje kůži.
- Polymyozitida a dermatomyozitida spojená se zhoubnými novotvary se může vyskytnout u pacientů jakéhokoli věku; jejich vývoj je nejčastěji pozorován u starších pacientů, stejně jako u pacientů s jinými onemocněními pojivové tkáně. Vývoj zhoubných novotvarů lze pozorovat jak 2 roky před, tak i 2 roky po vzniku myozitidy.
- Dětská polymyozitida nebo dermatomyozitida je spojena se systémovou vaskulitidou.
- Polymyozitida a dermatomyozitida se mohou vyskytnout i u pacientů s jinými onemocněními pojivové tkáně, nejčastěji s progresivní systémovou sklerózou, smíšeným onemocněním pojivové tkáně a systémovým lupusem (SLE).
Zařazení myozitidy trupových svalů do skupiny polymyozitid je nesprávné, protože se jedná o samostatné onemocnění charakterizované klinickými projevy podobnými chronické idiopatické polymyozitidě. Vyvíjí se však ve stáří, často postihuje svaly distálních částí těla (například horních a dolních končetin), má delší trvání, hůře reaguje na léčbu a je charakterizována typickým histologickým obrazem.
Diagnóza dermatomyozitidy a polymyozitidy
Polymyozitidu je třeba předpokládat u pacientů se stížnostmi na slabost proximálních svalů, s citlivostí nebo bez ní. Vyšetření na dermatomyozitidu je nutné u pacientů se stížnostmi na vyrážku připomínající heliotrop nebo Gottronův příznak, stejně jako u pacientů s projevy polymyozitidy v kombinaci s jakýmikoli kožními lézemi odpovídajícími dermatomyozitidě. Klinické projevy polymyozitidy a dermatomyozitidy se mohou podobat projevům systémové sklerózy nebo méně často systémového lupusu erythematosus (SLE) nebo vaskulitidy. Jistotu diagnózy zvyšuje splnění co největšího počtu z následujících pěti kritérií:
- slabost proximálních svalů;
- charakteristické kožní vyrážky;
- zvýšená aktivita enzymů svalové tkáně (kreatinkinázy nebo, pokud nedojde ke zvýšení její aktivity, aminotransferáz nebo aldolázy);
- charakteristické změny v myografii nebo MRI;
- charakteristické histologické změny v biopsii svalové tkáně (absolutní kritérium).
Svalová biopsie může vyloučit některé klinicky podobné stavy, jako je myozitida trunčních svalů a rabdomyolýza v důsledku virové infekce. Změny odhalené histologickým vyšetřením se mohou lišit, ale typické jsou chronický zánět, ložiska svalové degenerace a regenerace. Před zahájením potenciálně toxické léčby je nezbytná přesná diagnóza (obvykle histologickým ověřením). MRI dokáže detekovat ložiska edému a zánětu ve svalech a následně provést jejich cílenou biopsii.
Laboratorní vyšetření mohou potvrdit nebo naopak vyloučit podezření na přítomnost onemocnění a jsou také užitečná při posouzení jeho závažnosti, možnosti kombinace s jinou podobnou patologií a diagnostice komplikací. Navzdory skutečnosti, že u některých pacientů jsou detekovány antinukleární protilátky, je tento jev typičtější pro jiná onemocnění pojivové tkáně. Asi 60 % pacientů má protilátky proti antigenu jader (PM-1) nebo celých buněk brzlíku a Jo-1. Úloha autoprotilátek v patogenezi onemocnění zůstává nejasná, ačkoli je známo, že protilátky proti Jo-1 jsou specifickým markerem antisyntetázového syndromu, včetně fibrotizující alveolitidy, plicní fibrózy, artritidy a Raynaudovy choroby.
Pravidelné sledování aktivity kreatinkinázy je užitečné pro sledování léčby. U pacientů s těžkou svalovou atrofií však může být aktivita enzymu normální i přes přítomnost chronické aktivní myozitidy. MRI, svalová biopsie nebo zvýšená aktivita kreatinkinázy jsou často užitečné při rozlišení mezi relapsem polymyozitidy a myopatií indukovanou glukokortikoidy.
Vzhledem k tomu, že mnoho pacientů má nediagnostikované maligní nádory, někteří autoři doporučují screening všech dospělých s dermatomyozitidou a pacientů s polymyozitidou starších 60 let podle následujícího schématu: fyzikální vyšetření, včetně vyšetření prsou, pánve a konečníku (včetně testování stolice na okultní krvácení); kompletní krevní obraz; biochemie krve; mamografie; testování karcinoembryonálního antigenu; analýza moči; rentgen hrudníku. Někteří autoři zpochybňují potřebu takového screeningu u mladších pacientů, kteří nemají klinické známky malignity.
Co je třeba zkoumat?
Léčba dermatomyozitidy a polymyozitidy
Dokud se zánět nezmírní, měla by být fyzická aktivita omezena. Glukokortikoidy jsou léky první volby. V akutním stádiu onemocnění by měl být dospělým pacientům předepsán prednisolon (perorálně) v dávce 40 až 60 mg denně. Pravidelné stanovení aktivity kreatinkinázy je časným ukazatelem účinnosti: u většiny pacientů je pozorován pokles nebo normalizace během 6 až 12 týdnů po zvýšení svalové síly. Po normalizaci aktivity enzymů se dávka prednisolonu snižuje: nejprve přibližně o 2,5 mg denně po dobu jednoho týdne, poté rychleji; pokud dojde ke recidivě zvýšené aktivity svalových enzymů, dávka hormonu se opět zvyšuje. Uzdravení pacienti se obejdou bez glukokortikoidů, ale nejčastěji dospělí pacienti potřebují dlouhodobou léčbu glukokortikoidy (10–15 mg prednisolonu denně). Počáteční dávka prednisolonu pro děti je 30–60 mg/m2 jednou denně. Při remisi > 1 roku u dětí lze léčbu glukokortikoidy ukončit.
V některých případech se u pacientů užívajících vysoké dávky glukokortikoidů objeví náhlé zvýšení svalové slabosti, které může souviset s rozvojem glukokortikoidní myopatie.
V případě nedostatečné odpovědi na léčbu glukokortikoidy, stejně jako v případě rozvoje glukokortikoidní myopatie nebo jiných komplikací vyžadujících snížení dávky nebo vysazení prednisolonu, by měla být použita imunosupresiva (methotrexát, cyklofosfamid, azathioprin, cyklosporin). Někteří pacienti mohou dostávat pouze methotrexát (obvykle v dávkách přesahujících dávky používané k léčbě revmatoidní artritidy) po dobu delší než 5 let. Intravenózní imunoglobuliny mohou být účinné u pacientů refrakterních na farmakoterapii, ale jejich použití zvyšuje náklady na léčbu.
Myozitida spojená s primárními a metastatickými nádory, stejně jako myozitida svalů trupu, jsou obvykle méně refrakterní na léčbu glukokortikoidy. Remise myozitidy spojené se maligními nádory je možná po odstranění nádoru.
Jaká je prognóza dermatomyozitidy a polymyozitidy?
Dlouhodobá remise (a dokonce i klinické zotavení) po dobu 5 let je pozorována u více než poloviny léčených pacientů; u dětí je toto číslo vyšší. K relapsu však může dojít kdykoli. Celková pětiletá míra přežití je 75 %, u dětí vyšší. Příčinami úmrtí u dospělých jsou těžká a progresivní svalová slabost, dysfagie, snížená výživa, aspirační pneumonie nebo respirační selhání v důsledku plicních infekcí. Polymyozitida je závažnější a rezistentní na léčbu, pokud je dojde k poškození srdce a plic. U dětí může k úmrtí dojít v důsledku střevní vaskulitidy. Celková prognóza onemocnění je také určena přítomností maligních novotvarů.