Lékařský expert článku

Nové publikace
Syndrom Aper
Naposledy posuzováno: 04.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.

 [
[ Příčiny Syndrom Aper
Vývoj syndromu je vyvolán poruchou struktury genu umístěného na chromozomu 10. Tento gen je zodpovědný za proces oddělování prstů a navíc za včasné uzavření lebečních stehů.
Kromě toho se za příčiny patologie považují infekční onemocnění prodělaná během těhotenství (jako jsou zarděnky nebo tuberkulóza, syfilis nebo chřipka, stejně jako meningitida) a vystavení matky rentgenovému záření. Nejčastěji se takový syndrom vyskytuje u dětí narozených starším rodičům.
Patogeneze
Apertův syndrom je dědičný. Jeho typ je autozomálně dominantní (tj. v rodině, kde je jeden z budoucích rodičů nemocný, je pravděpodobnost narození dítěte s touto nemocí 50–100 %).
Unikátní mutace v receptoru 2 fibroblastového růstového faktoru (FGFR2) vede ke zvýšenému počtu progenitorových buněk, které se vyvíjejí podél osteogenní dráhy. To nakonec vede ke zvýšené tvorbě subperiostální kostní matrix a předčasné osifikaci lebečních stehů během fetálního vývoje. Pořadí a rychlost tavení stehů určuje stupeň deformity a postižení. Jakmile se šicí materiál zhojí, růst dalších tkání kolmých k tomuto stehu je omezen a srostlé kosti fungují jako jediná kostní konstrukce.
Prvním genetickým důkazem, že syndaktylie u Apertova syndromu je způsobena defektem receptoru pro keratinocytový růstový faktor (KGFR), bylo pozorování korelace mezi expresí KGFR ve fibroblastech a závažností syndaktylie.
Amblyopie a strabismus jsou častější u pacientů s mutací FGFR2 Ser252Trp a atrofie optického disku je častější u pacientů s mutací FGFR2 Pro253Arg. Pacienti s mutacemi FGR2 Ser252Trp mají významně vyšší prevalenci zrakového postižení ve srovnání s pacienty s mutací FGFR2 Pro253Arg.
Symptomy Syndrom Aper
Některé projevy onemocnění jsou jasně patrné již při narození dítěte, protože se vyvíjejí v matčině děloze. Mezi hlavní příznaky syndromu patří:
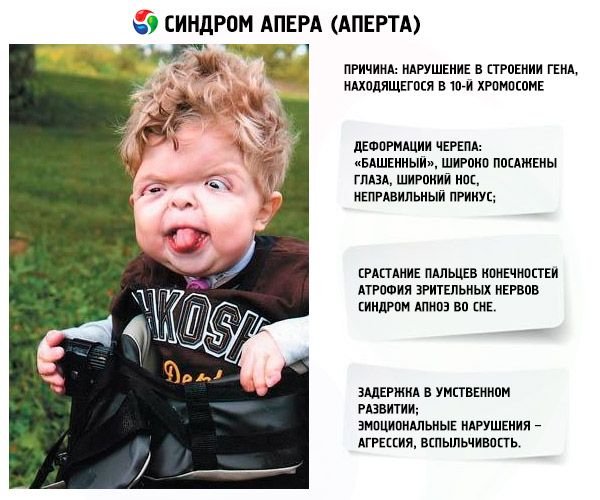
- Lebka je deformovaná - protahuje se do výšky a získává „věžovitý“ vzhled; navíc jsou oči široce posazené a mírně vypoulené (protože se zmenšuje velikost očních důlků); nos se rozšiřuje a vytváří se nesprávný skus (horní zuby nadměrně vyčnívají);
- Prsty končetin jsou zcela srostlé (hlavně prsteníček, stejně jako prostředníček a ukazováček) a vypadají jako kožní membrána nebo úplné srůstání kostí; navíc mohou narůstat další prsty;
- Mentální retardace (nevyskytuje se u každého);
- Atrofie optických nervů, v důsledku čehož se zraková ostrost snižuje (v některých případech dochází k úplné ztrátě zraku);
- Zvýšený nitrolební tlak, ke kterému dochází v důsledku předčasného uzavření lebečních stehů, se projevuje bolestmi hlavy a zvracením s nevolností;
- Protože horní čelist zůstává nedostatečně vyvinutá, vznikají dýchací potíže;
- Syndrom spánkové apnoe je častým jevem.
- Emoční projevy – agrese, nedostatek zdrženlivosti, silná popudlivost.
Komplikace a důsledky
Diagnostika Syndrom Aper
Pro stanovení diagnózy jsou nutné následující kroky:
- Lékař by měl analyzovat stížnosti pacienta a také jeho anamnézu. Je nutné zjistit, zda se v rodině vyskytly případy podobné patologie;
- Neurologické vyšetření k posouzení tvaru lebky a intelektuálního vývoje pacienta (speciální dotazníky a také rozhovor);
- Vyšetření fundusu za účelem zjištění přítomnosti příznaků zvýšeného intrakraniálního tlaku (otok optického disku a také rozmazání jeho okrajů);
- Pro posouzení stavu lebky se provádí rentgenový snímek;
- Počítačové a magnetické rezonanční zobrazování hlavy k vyšetření struktury mozku a lebky vrstvu po vrstvě, k určení přítomnosti příznaků předčasného srůstu lebečních švů a navíc hydrocefalu (v důsledku zvýšeného nitrolebního tlaku se hromadí přebytečný mozkomíšní mok (to je mozkomíšní mok, který podporuje metabolický proces a výživu mozku));
- Rentgenový snímek nohou a rukou k určení příčiny srůstu prstů (to je důležité pro plánování následného chirurgického zákroku);
- Může být předepsána konzultace s lékařským genetikem a neurochirurgem.
Testy
Provádí se genetická analýza běžných mutací vyskytujících se v genu typu FGFR2.
Diferenciální diagnostika
Tento syndrom je nutné odlišit od jiných genetických patologií, u kterých se kraniosynostóza pozoruje. Jedná se o onemocnění, jako jsou Pfeifferův, Crouzonův, Saethre-Chotzenův a Carpenterův syndrom. K vyloučení těchto anomálií se používají molekulárně genetické testovací metody.
Kdo kontaktovat?
Léčba Syndrom Aper
Chirurgie je považována za jedinou účinnou léčbu Apertova syndromu – pomáhá korigovat individuální fyzické vady a také koriguje mentální retardaci.
Během tohoto zákroku se uzavře koronální šev, aby se zabránilo možnému poranění mozku. Nejběžnější technikou je kraniofaciální distrakce, která zahrnuje stupňovitou trakci lebky. Ortodontická a/nebo ortognátní chirurgie se provádí k odstranění jednotlivých defektů obličeje.
Kromě toho pacienti podstupují chirurgickou korekci fúze prstů.
Reference
Lékařská genetika. Národní průvodce. Editoval Ginter EK, Puzyrev VP GEOTAR-Media. 2022