Nové publikace
Nové poznatky přispívají k lepšímu pochopení příčin Rettova syndromu
Naposledy posuzováno: 02.07.2025

Veškerý obsah iLive je lékařsky zkontrolován nebo zkontrolován, aby byla zajištěna co největší věcná přesnost.
Máme přísné pokyny pro získávání zdrojů a pouze odkaz na seriózní mediální stránky, akademické výzkumné instituce a, kdykoli je to možné, i klinicky ověřené studie. Všimněte si, že čísla v závorkách ([1], [2] atd.) Jsou odkazy na tyto studie, na které lze kliknout.
Pokud máte pocit, že některý z našich obsahů je nepřesný, neaktuální nebo jinak sporný, vyberte jej a stiskněte klávesu Ctrl + Enter.

Rettův syndrom je vzácná neurovývojová porucha, na kterou v současné době neexistuje žádný lék ani dobrá léčba. Způsobuje závažné fyzické a kognitivní příznaky, z nichž mnohé se překrývají s poruchami autistického spektra.
Rettův syndrom je způsoben mutacemi v genu MECP2, který je v mozku vysoce exprimován a zdá se, že hraje důležitou roli v udržování zdraví neuronů. Gen se nachází na chromozomu X a syndrom postihuje především dívky. Aby mohli vědci vyvinout léčbu Rettova syndromu, chtějí lépe porozumět genu MECP2 a jeho funkcím v mozku.
Výzkumníci, včetně spoluzakladatele Whiteheadova institutu Rudolfa Jaenische, studují gen MECP2 již desítky let, přesto mnoho základních faktů o tomto genu zůstává neznámých. Protein kódovaný genem MECP2 se podílí na regulaci genů; váže se na DNA a ovlivňuje hladiny exprese různých dalších genů nebo množství proteinu, které produkují.
Vědci však neměli úplný seznam genů ovlivněných MECP2 a neexistovala shoda v tom, jak MECP2 tyto geny ovlivňuje.
První studie MECP2 naznačovaly, že se jedná o represor, který snižuje expresi cílových genů, ale výzkum Jaenische a dalších již dříve ukázal, že MECP2 působí také jako aktivátor, který zvyšuje expresi svých cílů – a že by aktivátorem vůbec mohl být. Neznámý byl také mechanismus účinku MECP2, ani co přesně tento protein dělá pro změny v genové expresi.
Technologická omezení zabránila vědcům v získání jasných informací o těchto otázkách. Yanish, postdoktorand Yi Liu z jeho laboratoře a bývalý člen Yanishovy laboratoře Anthony Flamier, nyní odborný asistent ve výzkumném centru CHU Sainte-Justine na Univerzitě v Montréalu, však využili nejmodernější techniky k zodpovězení těchto zbývajících otázek ohledně MECP2 a získali nové poznatky o jeho roli ve zdraví a onemocněních mozku.
Jejich výsledky byly publikovány v časopise Neuron a vědci také vytvořili online úložiště svých dat MECP2, portál MECP2-NeuroAtlas, jako zdroj pro další výzkumníky.
„Myslím, že tento článek zásadně změní chápání lidí o tom, jak MECP2 způsobuje Rettův syndrom. Máme zcela nové chápání mechanismu a může poskytnout nové cesty pro vývoj léčby této nemoci,“ říká Janisch, který je také profesorem biologie na MIT.
Hlubší pochopení MECP2 v mozku
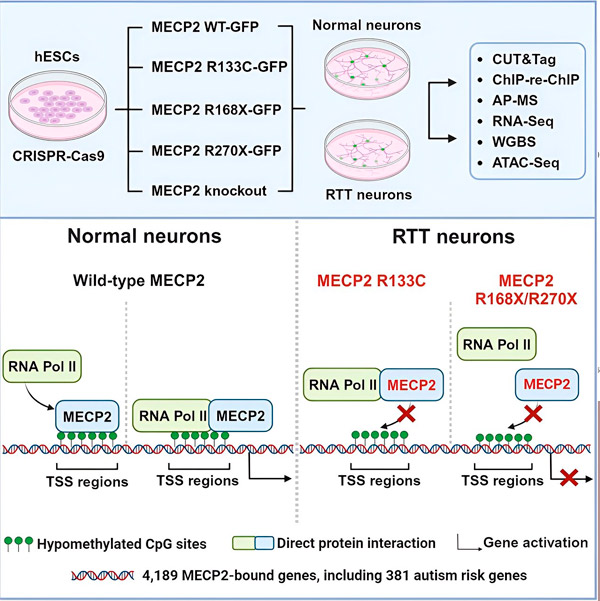
Vědci nejprve vytvořili podrobnou mapu, kde se MECP2 váže v genových sekvencích lidských neuronů, ať už v rámci genů nebo v regulačních oblastech DNA v jejich blízkosti. Použili přístup zvaný CUT&Tag, který dokáže s vysokou přesností přesně určit interakce proteinů s DNA.
Vědci objevili více než 4 000 genů spojených s MECP2. Zopakovali mapování v neuronech s běžnými mutacemi MECP2 spojenými s Rettovým syndromem, aby určili, kde je MECP2 v průběhu onemocnění vyčerpán.
Znalost genů, na které se MECP2 váže, umožnila Liu a Flamierovi začít navazovat souvislosti mezi cílovými oblastmi MECP2 a zdravím mozku. Zjistili, že mnoho z těchto cílových oblastí se podílí na vývoji a funkci neuronálních axonů a synapsí.
Také porovnali svůj seznam cílů MECP2 s databází genů asociovaných s autismem od Simons Foundation Autism Research Initiative (SFARI) a zjistili, že 381 genů v této databázi jsou cíli MECP2.
Zdroj: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Tato zjištění by mohla pomoci objasnit mechanismy, které jsou základem autistických symptomů u Rettova syndromu, a poskytnout dobrý výchozí bod pro zkoumání možné role MECP2 v autismu.
„Vytvořili jsme první integrovanou mapu epigenomu MECP2 ve zdraví a nemoci a tato mapa může být vodítkem pro budoucí výzkum,“ říká Liu. „Vědomí si, které geny jsou cílem MECP2 a které geny jsou při onemocnění přímo narušeny, poskytuje solidní základ pro pochopení Rettova syndromu a kladení otázek ohledně genové regulace v neuronech.“
Vědci se také zabývali tím, zda MECP2 zvyšuje nebo snižuje expresi svých cílových genů. V souladu s historií, kdy byl MECP2 identifikován některými jako aktivátor a jinými jako represor, Liu a Flamier našli příklady, kde MECP2 hrál obě role.
Zatímco MECP2 je častěji považován za represor, Liu a Flamier zjistili, že se jedná převážně o aktivátor – což potvrzuje předchozí zjištění Jaenische a Liu. Jeden nový experiment ukázal, že MECP2 aktivuje nejméně 80 % svých cílů, a jiný zjistil, že aktivuje až 88 % svých cílů.
Mapa cílových genů, kterou vědci vytvořili, poskytla další vhled do role MECP2 jako aktivátoru. Zjistili, že geny, které MECP2 aktivuje, se obvykle vážou na oblast DNA před genem, která se nazývá místo zahájení transkripce.
Toto je místo, kde buněčný aparát zahajuje proces transkripce genu do RNA, po kterém je RNA přeložena do funkčního proteinu, který je produktem genové exprese. Přítomnost MECP2 v místě zahájení transkripce, kde začíná genová exprese, je v souladu s jeho rolí jako aktivátoru genu.
Vědci se poté vydali na cestu k určení role, kterou hraje MECP2 v aktivaci genů. Zkoumali, na jaké molekuly se MECP2 v tomto místě váže, kromě DNA, a zjistili, že MECP2 interaguje přímo s proteinovým komplexem zvaným RNA polymeráza II (RNA Pol II). RNA Pol II je klíčový buněčný stroj, který přepisuje DNA do RNA. RNA Pol II nedokáže sama o sobě najít geny, takže k jejímu výkonu potřebuje řadu kofaktorů neboli proteinových spolupracovníků.
Vědci se domnívají, že MECP2 slouží jako jeden z takových kofaktorů, který pomáhá RNA Pol II zahájit transkripci v genech, kde se MECP2 váže. Strukturní analýza MECP2 identifikovala části molekuly, které se vážou na RNA Pol II, a další experimenty potvrdily, že ztráta MECP2 snižuje přítomnost RNA Pol II v příslušných místech zahájení transkripce, stejně jako hladiny exprese cílových genů.
To naznačuje, že Rettův syndrom může být způsoben sníženou transkripcí genů cílených proteinem MECP2 v důsledku mutací genu MECP2, které mu brání ve vazbě na RNA Pol II nebo na DNA. V souladu s touto myšlenkou jsou nejčastějšími mutacemi MECP2 spojenými s onemocněním zkrácení: mutace, při kterých chybí část proteinu, což může změnit interakci mezi MECP2 a RNA Pol II.
Vědci doufají, že jejich zjištění nejen změní naše chápání MECP2, ale že hlubší a širší pochopení toho, jak MECP2 ovlivňuje vývoj a funkci mozku, by mohlo vést k novým poznatkům, které pomohou lidem s Rettovým syndromem a souvisejícími poruchami, včetně autismu.
„Tento projekt je skvělým příkladem kolaborativní povahy Janischovy laboratoře,“ říká Flamier. „S Rudolfem jsme měli specifický problém související s Rettovým syndromem a já jsem měl zkušenosti s technologií CUT&Tag, která by tento problém mohla vyřešit. Během diskuse jsme si uvědomili, že bychom mohli spojit naše úsilí a nyní máme skvělé úložiště informací o MECP2 a jeho souvislostech s onemocněním.“